Using humanized liver mice to advance the development of LNP delivery systems
Nucleic Acid Insights 2026; 3(8), 233–242
DOI: 10.18609/nai.2026.030
Non-viral delivery systems, such as lipid nanoparticles (LNP), are driving the next wave of nucleic acid therapeutics by enabling efficient intracellular delivery of mRNA and siRNA, including gene-editing payloads in clinical settings. LNP offer scalable manufacturing, tunable biodistribution, and favorable safety profiles compared with viral vectors, making them a cornerstone of next-generation therapeutics. Although LNP delivery is the basis for a number of clinically approved products, challenges remain for enabling future LNP delivery systems. One critical factor is the identification of appropriate preclinical models to test the efficacy and toxicity of LNP-based therapeutics at an early stage of development, as this can have a big impact on the chances of the therapeutic making it to market.
How to reach your target? LNP & beyond
Current LNP represent the culmination of decades of innovation in lipid chemistry and nanoparticle engineering. Modern LNP typically consist of ionizable lipids, helper phospholipids, cholesterol, and polyethylene glycol (PEG)–lipids, each contributing to stability, cellular uptake, and endosomal escape. Advances in ionizable lipid design have been particularly impactful, enabling efficient nucleic acid encapsulation at low pH, reduced toxicity at physiological pH and enhanced endosomal release of mRNA to the cytoplasm [1][2][3].
Beyond LNP, the non-viral delivery landscape continues to diversify. Novel lipid chemistries, biodegradable and stimuli-responsive ionizable lipids, cationic polymers such as polyethylenimine (PEI), lipid–polymer hybrids, oligosaccharides, inorganic nanoparticles, and ligand-targeted systems are actively being explored to improve tissue specificity, intracellular trafficking, and tolerability [4]. While these next-generation platforms hold promise, their increasing complexity also amplifies the challenge of predicting in vivo behavior and clinical responses.
Species specificity in liver targeting
LNP preferentially target the liver, but the process of entering the hepatocyte still involves multiple stages, including:
- Protein corona formation
- Hepatic sinusoidal transport
- Receptor-mediated uptake
- Endosomal escape, and
- Intracellular trafficking, which collectively determine delivery efficiency and therapeutic potency (Figure 1) [3][5]
| Figure 1. Species specific differences in LNP uptake into hepatocytes. |
|---|
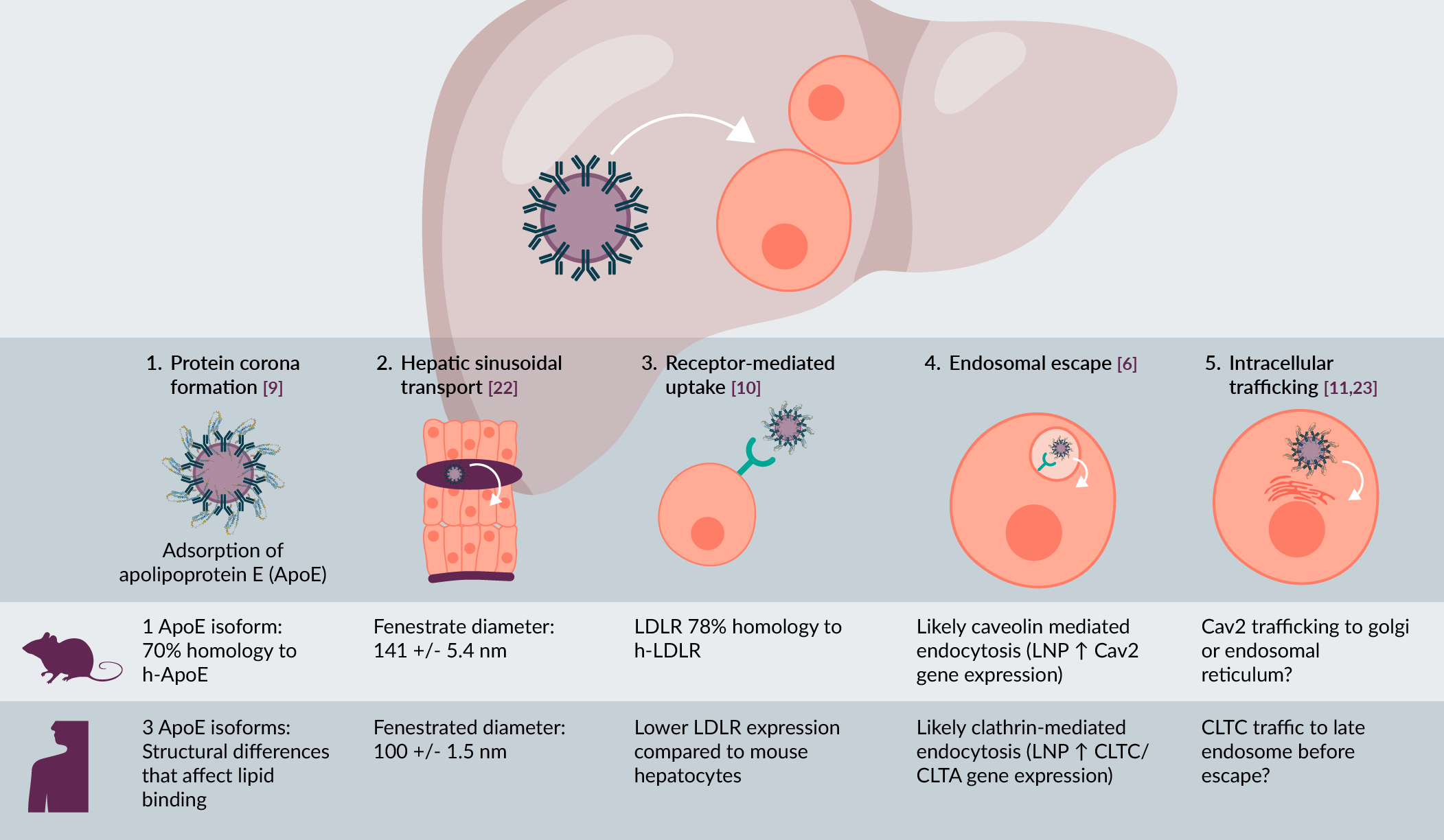 |
| Data from [6][9][10][11][22][23]. |
Importantly, each of these steps is highly influenced by species-specific differences in liver physiology and hepatocyte biology [6]. For example, the binding of apolipoprotein E (ApoE) to LNP in the blood stream, followed by uptake of these particles via interaction with the low-density lipoprotein (LDL) receptor is a key pathway for uptake of LNP by hepatocytes. Although the pathway is conserved between rodents and humans, substantial differences in ApoE expression levels, isoform distribution, and receptor interactions can lead to divergent uptake kinetics and biodistribution profiles [7][8]. Beyond uptake, interspecies differences in endosomal maturation pathways, lipid metabolism, serum factors, and innate immune sensing further complicate the translation of LNP potency and tolerability from preclinical models to patients [6][9][10][11][12]. Although mice are often used to screen for the best LNP to take forward in clinical development, efficient delivery in mice does not always translate well to efficient delivery in non-human primates (NHP), or humans [13]. Thus, the lack of preclinical models that recapitulate these human-specific mechanisms has been a major bottleneck in LNP therapeutic development.
PXB-mice and other humanized liver chimeric mouse models, which have functional human hepatocytes that repopulate the liver, could help to mitigate this problem as they recapitulate key features of human liver physiology and metabolism. PXB-mice are generated on a cDNA-uPA/SCID (severe combined immunodeficiency) background, in which the controlled expression of the urokinase-type plasminogen activator (uPA) transgene in the murine hepatocytes results in chronic murine hepatotoxicity and an environment conducive to high levels of repopulation with human hepatocytes [14]. Typically, transplanted animals have livers that are 85–95% human hepatocytes, with normal liver architecture and validated activity of human Cytochrome P450 (CYP) enzymes, liver transporters and more [15][16][17][18]. Moreover, they have uniform engraftment of human hepatocytes throughout all the lobes of the liver and have similar ‘metabolic zonation’ as human liver tissue [19][20]. Additionally, they have a human lipoprotein profile [21]. Thus, they could be used to evaluate delivery efficiency, expression levels, and tolerability, directly in human cells, at an earlier stage of therapeutic development. As non-viral delivery technologies continue to advance, aligning nanoparticle design with human biology as early as possible will be essential to fully realizing their therapeutic potential. In this article, we discuss how humanized mouse models could play an important role in this space, by providing a platform that can overcome the divide between in vitro cell models and the clinic.
Efficient delivery of LNP is observed in the PXB-mouse liver
To validate the utility of humanized liver mouse models in the characterization of LNP for therapeutic uses, a recent study examined LNP delivery of mRNA in PXB-mice to assess the distribution, efficiency, and tolerability of an LNP formulation developed by Acuitas Therapeutics [24–27]. In this study, PXB-mice were injected intravenously with an LNP encapsulating a reporter Immunoglobulin G (IgG) mRNA at different dose levels. Delivery and expression in both human and mouse hepatocytes in the PXB-mouse liver was evident from immunohistochemistry (IHC) staining for IgG protein and a human-specific marker in serial, adjacent sections (Figure 2).
| Figure 2. IHC and hematoxylin-eosin (H&E) staining of liver sections from PXB-mice dosed with LNP at 3.0 mg/kg. |
|---|
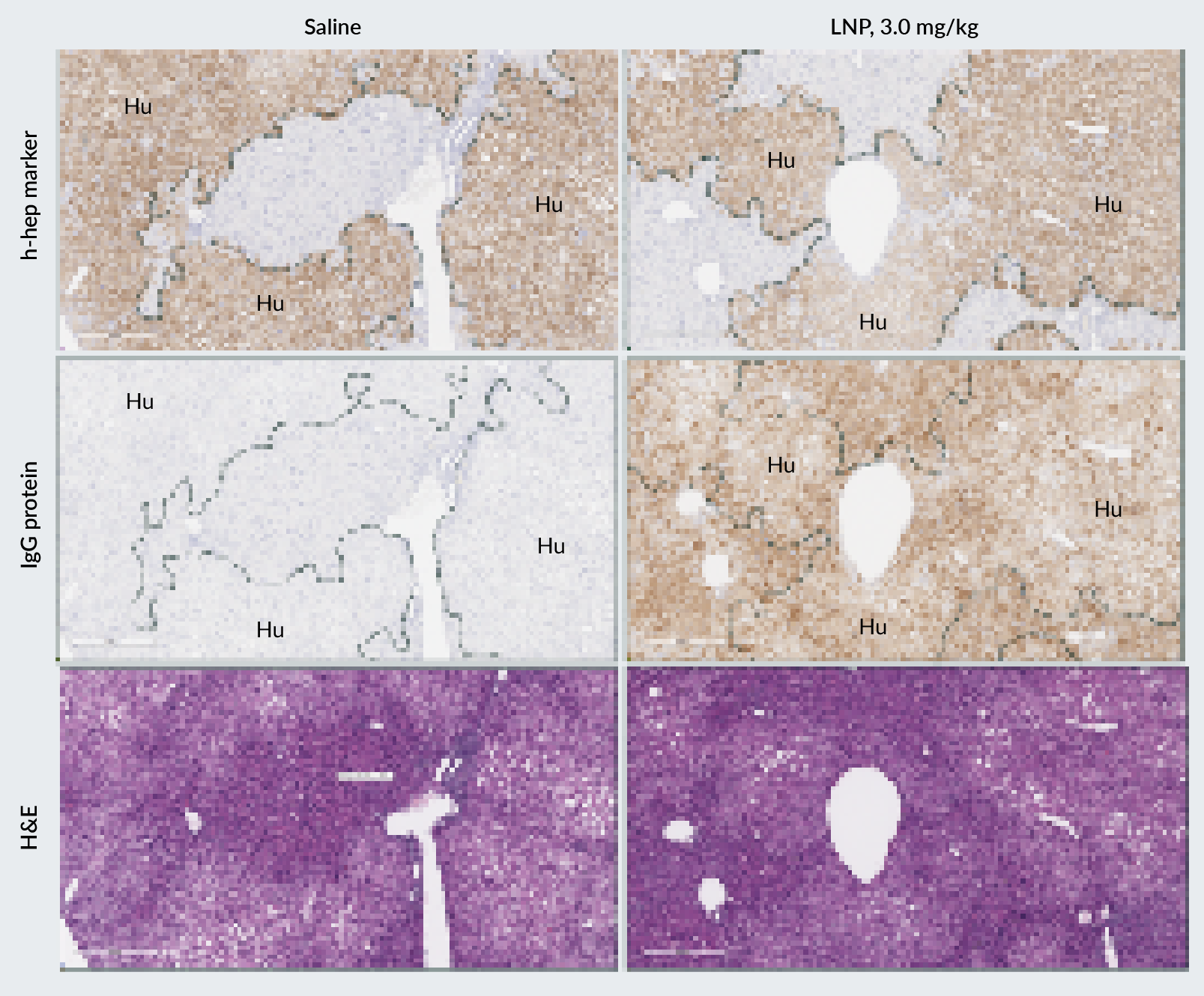 |
| Samples were collected 24-hours post-LNP administration. IgG protein expression (LNP payload) was observed in regions of human hepatocytes (identified by the expression of a human-specific hepatocyte marker). |
We next assessed serum IgG levels to determine LNP potency. A clear dose-dependent response was observed at 24 hours post-dosing (Figure 3A). While blood biochemistry analysis demonstrated dose-dependent increases in total alanine transaminase (ALT), aspartate aminotransferase (AST), and human ALT1 (hALT1), no changes were observed for serum total bilirubin levels (TBIL) across all dose levels (Figure 3B). This suggests the LNP may cause mild hepatocellular stress but that overall bile flow and liver functioning are not impaired [28]. Consistent with this, no clinical manifestations were observed even at the highest dose level tested (5 mg/kg), indicating that the LNP formulation was well tolerated by PXB-mice.
| Figure 3. Serum IgG expression (LNP payload) and blood biochemistry markers were assessed to determine LNP potency and safety. |
|---|
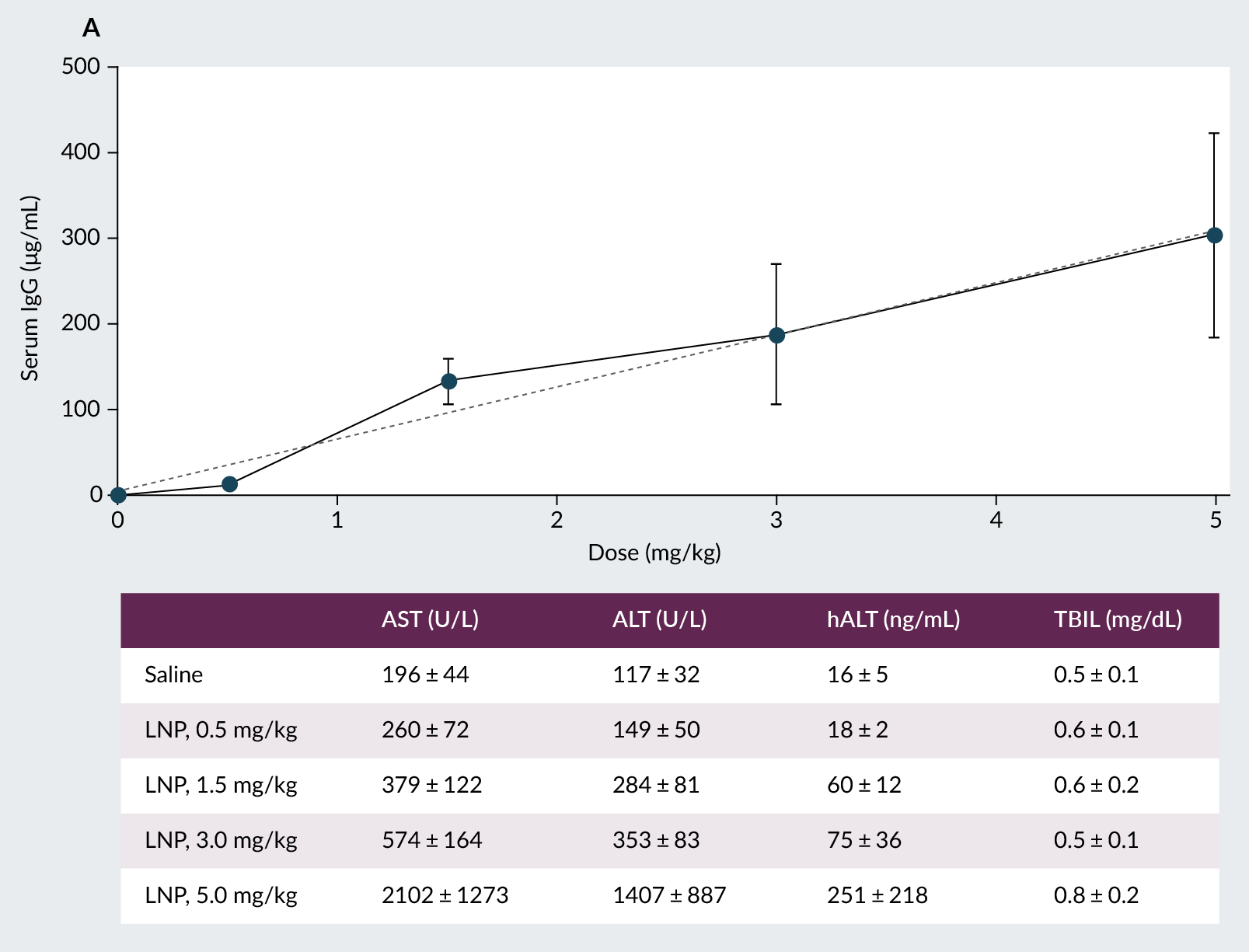 |
| PXB-mice were intravenously dosed with 0.5 mg/kg, 1.5 mg/kg, 3.0 mg/kg or 5.0 mg/kg. A: Serum IgG protein and total ALT, human-specific ALT1, AST, and B: total bilirubin levels were assessed at 24-hours post-dose. |
While human hepatocytes comprise the majority of the liver of the PXB-mouse, there can be 5–15% residual murine hepatocytes present in a typical animal used in a study. Yamazaki et al. recently carried out a study to confirm that human-specific targeting and silencing of the desired gene is possible. PXB-mice were injected with an LNP encapsulating an siRNA designed to knock down the expression of transthyretin (Ttrl/TTR) [29]. This LNP contains an asymmetric ionizable lipid and has been previously shown to be effective in both mice and non-human primates (NHP) [30]. Transthyretin is primarily expressed by hepatocytes and is secreted into the bloodstream allowing for straightforward in vivo monitoring of knockdown efficiency. This work showed that species-specific targeting is feasible as there was a significant reduction in plasma human TTR levels observed at 1-, 3-, and 7-days post-injection with siRNA targeting human TTR (hTTR)-LNP [29]. Additionally, hepatic hTTR mRNA levels were significantly reduced in a dose-dependent manner whereas mouse TTR protein levels were not affected.
Although delivery efficiency within humanized mice livers is likely to be LNP-dependent, the two examples above indicate that clinically-optimized LNP can be well-tolerated and extensively distributed throughout the PXB-mouse liver, with no preferential delivery between human and mouse hepatocytes. Taken together, these studies indicate that LNP can be used for various applications in the PXB-mouse, including mRNA and oligonucleotide delivery.
The PXB-mouse: improved prediction of RNA therapeutic toxicity
Toxicity is a key reason for the clinical failure of many therapeutics. Frequently preclinical models are not sufficient to detect human-specific toxicity, mainly due to species-specific differences in metabolism and physiology. Humanized liver mice can provide insights into potential safety issues at an earlier stage of therapeutic testing due to the presence of human liver metabolic enzymes and transporters. The PXB-mouse has previously been shown to more effectively predict human hepatotoxicity than conventional rodent and NHP models. Examples include the small molecule, troglitazone [31], and a monoclonal antibody therapeutic, KMTR2 [32]. Furthermore, the liver of the PXB-mouse has a human-specific transcriptome, which is important for understanding whether oligonucleotide therapeutics have off-target effects. For example, an siRNA designed to target hepatitis B virus (HBV) transcripts, ALN-HBV, significantly increased human ALT levels in a dose dependent manner in PXB-mice and clinical trial participants [33]. While this siRNA was conjugated with N-acetylgalactosamine (GalNAc), similar compounds were not associated with liver enzyme elevations; therefore, the observed increase in liver enzyme was likely due to off-target effects of the siRNA sequence. As such, ALN-HBV was modified, and the modified version, VIR-2218, effectively reduced viral infection levels in HBV-infected PXB-mice without ALT elevations [33]. Importantly, PXB-mice recapitulate clinical findings where other preclinical models (rats and NHP) failed to predict human-specific toxicity of ALN-HBV [34].
The PXB-mouse has also been used to de-risk LNP delivery systems. In a recent HBV study, a hetero-gapmer targeting a host factor (dedicator of cytokinesis 11; DOCK11) was used in conventional mice transduced with an (adeno-associated virus) AAV-HBV construct to model HBV. This treatment was well tolerated and effective in suppressing HBV in this model. However, when tested in HBV-infected PXB-mice, no changes were observed for HBV parameters and there were signs of liver toxicity observed in both the DOCK11-gapmer and in the gapmer control animals. Gapmers have previously been shown to cause more hepatotoxicity than siRNA therapeutics in a hepatic ribonuclease H1 (RNase H1) dependent manner [35]. Thus, due to the observed human-specific hepatotoxicity, the authors switched approach and generated an LNP-encapsulated siRNA targeting DOCK11. They found that in HBV-infected PXB-mice treated with LNP-siRNA DOCK11 there was a significant reduction in HBV DNA and covalently closed circular DNA (cccDNA) levels [36]. Unlike gapmer treatment, the LNP-siRNA had no effect on body weight or human albumin levels demonstrating that this therapeutic was well tolerated. Taken together, the PXB-mouse model is valuable for predicting toxicity of RNA therapeutics, including those delivered via LNP.
LNP delivery in PXB-mice compares to non-human primates
Non-human primate models are regularly used to predict LNP delivery and efficiency in humans. However, they come at a high cost, both financially and ethically. Furthermore, although NHPs are genetically similar to humans, slight genetic differences may reduce the ability to predict efficacy for certain human-specific genes. Therefore, having a more cost-effective model that predicts efficiency and safety for human-specific LNP therapeutics is important. Humanized liver mice can bridge this gap. For example, a study by Jain et al. in 2018 showed similar protein suppression was observed in PXB-mice and NHPs following LNP delivery of a microRNA (miR) target site (miRts) for miR122, demonstrating that PXB-mice can be used for preclinical screening of LNP therapeutics [37].
Conclusions
Overall, the data discussed here indicate that humanized liver mouse models, such as the PXB-mouse, can be used to assess LNP delivery, safety, and efficacy. The ability to test in vivo delivery to human hepatocytes, to target human genes and evaluate pharmacodynamic activity, and to detect human-specific hepatotoxicity during preclinical development can provide unique insights into the expected behavior of LNP delivery systems in a clinical setting. While these models have limitations, including residual mouse hepatocytes and murine non-parenchymal cells, humanized liver mouse models should be considered as valuable additions to preclinical pipelines, to identify the most promising LNP for clinical application.
References
1. Semple SC, Klimuk SK, Harasym TO, et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta 2001; 1510, 152–166. Crossref
2. Semple SC, Akinc A, Chen J, et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010; 28(2), 172–176. Crossref
3. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021; 6(12), 1078–1094. Crossref
4. Wang J, Ding Y, Chong K, et al. Recent advances in lipid nanoparticles and their safety concerns for mRNA delivery. Vaccines 2024; 12(10), 1148. Crossref
5. Kulkarni JA, Cullis PR, van der Meel R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther. 2018; 28(3), 146–157. Crossref
6. Hatit MZC, Lokugamage MP, Dobrowolski CN, et al. Species-dependent in vivo mRNA delivery and cellular responses to nanoparticles. Nat. Nanotechnol. 2022; 17(3), 310–318. Crossref
7. Akinc A, Maier MA, Manoharan M, et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019; 14(12), 1084–1087. Crossref
8. Sago CD, Krupczak BR, Lokugamage MP, Gan Z, Dahlman JE. Cell subtypes within the liver microenvironment differentially interact with lipid nanoparticles. Cell. Mol. Bioeng. 2019; 12(5), 389–397. Crossref
9. Lewandowski CT, Maldonado Weng J, LaDu MJ. Alzheimer’s disease pathology in APOE transgenic mouse models: the who, what, when, where, why, and how. Neurobiol. Dis. 2020; 139, 104811. Crossref
10. Minniti ME, Pedrelli M, Vedin LL, et al. Insights from liver-humanized mice on cholesterol lipoprotein metabolism and LXR-agonist pharmacodynamics in humans. Hepatology 2020; 72(2), 656–670. Crossref
11. Narayana YV, Gadgil C, Mote RD, Rajan R, Subramanyam D. Clathrin-mediated endocytosis regulates a balance between opposing signals to maintain the pluripotent state of embryonic stem cells. Stem Cell Rep. 2019; 12(1), 152–164. Crossref
12. Stone D, Takeuchi R, Dulin H, et al. Serum factors create species-specific barriers to hepatic gene transfer by lipid nanoparticles in liver-humanized mice. Mol. Ther. Methods Clin. Dev. 2025; 33(2), 101470. Crossref
13. Zenhausern R, Jang B, Schrader Echeverri E, et al. Lipid nanoparticle screening in nonhuman primates with minimal loss of life. Nat. Biotechnol. 2025; published online Jun 26. Crossref
14. Tateno C, Kawase Y, Tobita Y, et al. Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice. PLoS One 2015; 10(11), e0142145. Crossref
15. Tateno C, Yoshizane Y, Saito N, et al. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 2004; 165(3), 901–912. Crossref
16. Tateno C, Miya F, Wake K, et al. Morphological and microarray analyses of human hepatocytes from xenogeneic host livers. Lab. Invest. 2013; 93(1), 54–71. Crossref
17. Miyamoto M, Iwasaki S, Chisaki I, Nakagawa S, Amano N, Hirabayashi H. Comparison of predictability for human pharmacokinetics parameters among monkeys, rats, and chimeric mice with humanised liver. Xenobiotica 2017; 47(12), 1052–1063. Crossref
18. Feng B, Liang G, Zetterberg C, et al. Utility of chimeric mice with humanized livers for predicting hepatic organic anion-transporting polypeptide 1B-mediated clinical drug-drug interactions. Drug Metab. Dispos. 2024; 52(10), 1073–1082. Crossref
19. Tateno C, Kojima Y. Characterization and applications of chimeric mice with humanized livers for preclinical drug development. Lab. Anim. Res. 2020; 36, 2. Crossref
20. Sultana N, Izawa T, Kamei T, et al. Application of humanized mice to toxicology studies: properties of chimeric mice with humanized liver (PXB-mice) for hepatotoxicity. J. Toxicol. Pathol. 2025; 38(2), 183–189. Crossref
21. Papazyan R, Liu X, Liu J, et al. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J. Lipid Res. 2018; 59(6), 982–993. Crossref
22. Wisse E, Jacobs F, Topal B, Frederik P, De Geest B. The size of endothelial fenestrae in human liver sinusoids: implications for hepatocyte-directed gene transfer. Gene Ther. 2008; 15(17), 1193–1199. Crossref
23. Matthaeus C, Taraska JW. Energy and dynamics of caveolae trafficking. Front. Cell Dev. Biol. 2021; 8, 614472. Crossref
24. Ansell SM, Du X. Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids. WO2015199952A1. Dec 30, 2015.
25. Du X, Ansell SM. Lipids and lipid nanoparticle formulations for delivery of nucleic acids. WO2017004143A1. Jan 5, 2017.
26. Ansell SM, Du X. Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids. WO2017075531. May 4, 2017.
27. Du X. Lipids for lipid nanoparticle delivery of active agents. WO2020146805. Jul 16, 2020.
28. Thakur S, Kumar V, Das R, Sharma V, Mehta DK. Biomarkers of hepatic toxicity: an overview. Curr. Ther. Res. Clin. Exp. 2024; 100, 100737. Crossref
29. Yamazaki K, Kubara K, Sugahara G, et al. Species-specific gene expression manipulation in humanized livers of chimeric mice via siRNA-encapsulated lipid nanoparticle treatment. Mol. Ther. Methods Clin. Dev. 2025; 33(2), 101466. Crossref
30. Suzuki Y, Hyodo K, Suzuki T, Tanaka Y, Kikuchi H, Ishihara H. Biodegradable lipid nanoparticles induce a prolonged RNA interference-mediated protein knockdown and show rapid hepatic clearance in mice and nonhuman primates. Int. J. Pharm. 2017; 519, 34–43. Crossref
31. Kakuni M, Morita M, Matsuo K, et al. Chimeric mice with a humanized liver as an animal model of troglitazone-induced liver injury. Toxicol. Lett. 2012; 214(1), 9–18. Crossref
32. Nihira K, Nan-Ya KI, Kakuni M, et al. Chimeric mice with humanized livers demonstrate human-specific hepatotoxicity caused by a therapeutic antibody against TRAIL-receptor 2/death receptor 5. Toxicol. Sci. 2019; 167(1), 190–201. Crossref
33. Gane E, Lim YS, Kim JB, et al. Evaluation of RNAi therapeutics VIR-2218 and ALN-HBV for chronic hepatitis B: results from randomized clinical trials. J. Hepatol. 2023; 79(4), 924–932. Crossref
34. Schlegel MK, Janas MM, Jiang Y, et al. From bench to bedside: improving the clinical safety of GalNAc-siRNA conjugates using seed-pairing destabilization. Nucleic Acids Res. 2022; 50(12), 6656–6670. Crossref
35. Kasuya T, Hori S, Watanabe A, et al. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci. Rep. 2016; 6, 30377. Crossref
36. Okada H, Sakamoto T, Nio K, et al. Lipid nanoparticle-encapsulated DOCK11-siRNA efficiently reduces hepatitis B virus cccDNA level in infected mice. Mol. Ther. Methods Clin. Dev. 2024; 32(3), 101289. Crossref
37. Jain R, Frederick JP, Huang EY, et al. MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid Ther. 2018; 28(5), 285–296. Crossref
Affiliations
Amanda R Burmeister, Sara K Donnelly, and Matthew Baginski, PhoenixBio USA Corporation, New York, NY, USA
Hilda HT Au and Steven HY Fan, Acuitas Therapeutics, Vancouver, British Columbia, Canada
Authorship & conflict of interest
Contributions: The named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: Amanda R Burmeister, Sara K Donnelly, and Matthew Baginski all work for PhoenixBio, which has developed the PXB-mouse. Hilda HT Au and Steven HY Fan have no conflicts of interest to declare.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
AI process statement: No AI tools were used in the preparation of this article.
Article & copyright information
Copyright: Published by Nucleic Acid Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2026 PhoenixBio USA Corporation. Published by Nucleic Acid Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article was written by the named authors and reviewed by BioInsights’ Editorial team to ensure clarity, scientific accuracy, and alignment with BioInsights’ editorial standards. Where indicated, the article was externally peer reviewed.
Submitted for peer review: Apr 24, 2026.
Revised manuscript received: May 26, 2026.
Publication date: Jun 5, 2026.
![]()