
Downstream bioprocessing of AAV vectors: industrial challenges & regulatory requirements
Cell Gene Therapy Insights 2018; 4(2), 131-146.
10.18609/cgti.2018.016
With an increasing number of clinical trials and impressive results in the field of gene therapy, recombinant adeno-associated virus (rAAV) is confirmed as one of the most promising viral vectors. Five years after the European Medicines Agency approved Glybera, the recent marketing authorization of Luxturna by the Food and Drug Administation in the United States marks the concretization of years of vector development. It also highlights the urgency to set up industrial manufacturing processes to support economically feasible commercialization. While several expression systems coexist as upstream processes in rAAV manufacturing, the downstream part is more conserved between processes. However, as there are no established specifications for rAAV vector preparation purity, it is not clear how to determine whether a purification process meets the safety requirements. In addition, there are characteristics unique to rAAV, like the presence of empty viral particles, which represent a technical hurdle to develop standardized and scalable downstream process leading to consistent quality of the vector. This review describes the existing purification technologies and the way they address current regulatory requirements.
Approximately 10 years ago, gene therapy trials for Leber Congenital Amaurosis and Hemophilia B highlighted the enormous potential of adeno-associated virus (AAV) vectors for human gene therapy [1,2]. Since then, AAV has been evaluated in several clinical trials and numerous non-clinical studies. Recently, two public companies, AveXis and Audentes communicated on their websites clinical results on infants suffering from spinal muscular atrophy and myotubular myopathy, respectively [3,4] further demonstrating the potential of AAV as one of the most potent/promising vector to treat genetic disorders.
The utility of AAV vectors has been acknowledged by the European Medicine Agency (EMA) and the Food and Drug Administration (FDA) in the United States since two products received marketing approval, Glybera (uniQure) in EU in 2012 and Luxturna (Spark Therapeutics) in 2018 in the US [5,6].
Despite these successes, AAV vectorology remains suboptimal. Particularly, manufacturing sufficient quantities of rAAV to support commercial phase is one of the most urgent problems to be addressed. As a matter of fact, conventional AAV production processes show limited productivity and/or scalability resulting in high cost of goods, which translates in unprecedented high costs for the treatments. Luxturna treatment costs 850 000 $ and Glybera 1 000 000 €. Obviously, it is difficult to predict a bright future for gene therapy if the costs remain unaffordable for the patients.
Manufacturing processes for biologics are usually composed of two phases: the upstream process (USP) consisting of the production of active ingredient by a cell substrate and the downstream process (DSP) involving orthogonal methodologies for the removal of process impurities. For AAV, many efforts have been deployed in USP resulting in a variety of brilliant technologies that have been reviewed elsewhere [7, 8, 9, 10]. While further improvements are still awaited to increase the productivity in USP, the purification of AAV relies on methods that are more conventional. Nevertheless, DSP is subject to change as the regulatory guidelines evolve to become more stringent, and may take the first products approved for marketing as a benchmark or reference point. In addition, the presence of empty particles and non-product specific packaged DNA sill represent technical challenges that have not been fully addressed yet.
Purpose & challenges of AAV Downstream Processing
The purpose of downstream processing is to purify the active ingredient from the upstream process-derived impurities in such a way that the final product meets a level of purity that reduce risks to the patients safety. Over the years, several expression systems have been designed in an attempt to increase AAV productivity and cost effectiveness [7–10]. Currently, 4 major technologies are used in the upstream part of manufacturing processes:
- Transient transfection of HEK293 cells,
- Herpes simplex virus 1 (HSV-1) expression system principally used in BHK21 and HEK293 cells,
- Baculovirus expression vector system (BEVS) principally used in Sf9 insect cells,
- Adenovirus-induced stable cell lines, usually derived from HeLa cells
To some extent, the process impurities are similar as they consist essentially of DNA originating from host cells, helper virus or plasmids and proteins from the cells and the helper virus. Therefore, the DSP is not significantly altered by the USP. The only exception is the possible presence of infectious viruses depending on the expression system chosen (Table 1). When HSV-1, Baculovirus or Adenovirus are used upstream, additional steps must be undertaken to ensure their removal because of their potential immunogenicity, the risk of replication competent particles and pathogenicity for the patients [11,12].
| Table 1: AAV expression systems used in manufacturing and their related impurities to be eliminated by downstream processing. | ||||
|---|---|---|---|---|
| AAV production system | Transient Transfection | Adenovirus induction of stable cell line | HSV-1 expression system | Baculovirus expression system |
| Cell line frequently used | Human (HEK293) | Human (HeLa) | Hamster (BHK21) | Insect (Sf9) |
| Type of impurity to be eliminated by downstream processing | ||||
| Particulate cell debris | + | + | + | + |
| Proteins from cells or expression system | + | + | + | + |
| DNA from cells or expression system | + | + | + | + |
| Oncogenic sequences | + | + | ? | ? |
| Infectious virus from expression system | - | + | + | + |
| Other process-derived impurities (BSA, DNAse, detergents, leachables) | + | + | + | + |
For DSP experts, purification of AAV vectors may not appear as a challenge at first sight because AAV capsids are relatively small (icosahedra of approximately 20–25 nm diameter) and very stable compared to other viruses. These features are very significant for a purification process: on one hand, the vector can pass through many kinds of filters including 0.2 µm filters, which facilitates the control of product asepsis. On the other hand, it can resist – to a reasonable extent that needs to be defined accurately – a range of drastic/stringent conditions relating to shear stress, pH variation and high conductivity buffers.
However, a few characteristics related to AAV vector biology complicates the DSP. Firstly, many of the upstream processes can deliver vector titers between 1E4 to 5E5 vector genomes (vg)/cell, usually corresponding to volumetric titers between 1E10 and 2E11 vg/mL in the crude harvests [8, 9, 13, 14, 15]. The clinical doses of AAV are in the range of 1E12 vg/kg for liver gene therapy (Hemophilia B for instance) [2] to 1E14 vg/kg for neuromuscular disorders (Spinal Muscular Atrophy or Myotubular Myopathy) [3,4]. This means that the bulk drug substance has to be concentrated 100-fold to 10,000-fold to reach titers compatible with a reasonable administration volume (a few ml/kg for intravenous infusion). Such a level of concentration becomes a challenge in terms of purity: actually, even after a massive reduction of HCP and DNA, these residues may raise to unsafe titers after product concentration. For clinical/commercial manufacture, it is a challenge also because bioreactors operating at 200L to 500L require concentrating the final product to 100-200 mL. Very few devices are available to operate with such small volumes in GMP conditions (like sterile filtration systems or automated fill and finish instruments).
Another challenge associated with the purification of AAV vectors is the presence of empty capsids. During the upstream process, production of AAV particles lacking the vector DNA occurs to various levels depending on several factors like the genome length, the serotype and the expression system [8,15]. In some cases, empty particles can represent up to 90% of a clinical product [16]. Since full and empty particles are physically highly similar, differing principally by buoyant density, they cannot be discriminated by a standard downstream process. Specific technologies have to be used when there is a need to eliminate empty capsids.
Finally, AAV particles have been reported to contain DNA fragments derived from host cell DNA, helper virus and/or plasmid DNA. In the triple transfection system, the amount of particles containing HEK293 host cell DNA and plasmid DNA represents 0.3% to 3% and 0.8% to 5.3%, respectively, of the quantity of vector-containing capsids [17–19]. In BEVS, Sf9 host cell DNA and Baculovirus DNA are packaged in AAV capsids at a frequency of 0.03% and 2.1%, respectively [20]. Hauck et al. demonstrated using Southern blotting that the non-vector encapsidated DNA has variable length that can extend to 4.3 kb [18]. Technically, it seems impossible for a purification process to separate capsids containing the therapeutic genome from those containing illegitimate DNA fragments. This could represent a safety concern; however, Hauck et al. also showed that unspecifically encapsidated DNA could not drive mRNA expression in vitro and in vivo. This was demonstrated by monitoring the AAV cap gene – originating from the helper plasmid – which was detected at a frequency of 0.018% in a vector preparation. It was hypothesized that non-vector DNA was packaged as a single strand form [18,19]. Without the AAV inverted terminal repeats (ITRs) flanking the single strand DNA, no complementary strand can be synthesized after cell transduction. As such, ssDNA is expected to be unstable, which could explain the absence of transcription.
Quality & regulatory requirements
When the first clinical trials on AAV-based gene therapy started more than 20 years ago [21], there was no specific regulatory guidance for gene therapy products. The first vectors entering clinical phases were subjected to the guidelines for viral vaccines. This was appropriate for some aspects, especially for the management of viral safety and cell bank testing. As a matter of fact, any product manufactured in a cell substrate may be contaminated by adventitious agents. Therefore, the same rules apply to any kind of biologicals.
However, for a few other aspects, some of the standard requirements are difficult to meet. Particularly, the specifications for residual host cell DNA (10 ng/dose) were arbitrarily set and remain unrealistic to achieve for an AAV vector principally because of the presence of host cell DNA within the viral particles. Therefore, a certain degree of tolerance based on the risk/benefit balance for the patient has been allowed.
As a significant number of AAV products have entered clinical phases – with two of them having reached marketing authorization (Glybera and Luxturna) – more specific guidelines and more stringent requirements are expected from the Medicine Agencies worldwide. In Europe, the EMA has completed the Pharmacopeia by the monograph 5.14 dedicated to gene therapy products including AAV vectors [12], and the FDA has published a general guidance for Gene Therapy products [22].
These guidelines do not provide quantitative specifications for DNA, host cell and helper virus proteins or empty capsids. The acceptable levels are usually defined on a case-by-case study, depending on the pathology and on the total amounts of each impurity to be administered to the patients. Therefore, the manufacturers have to define their own product specifications on the basis of the risks associated with every type of impurity that is possibly present in the final product. For AAV vectors, the major points to consider are described below:
Originally, the regulatory levels for residual host cell DNA were based on the theoretical risk of oncogenicity. Particularly, continuous cell lines contain oncogenic sequences (the most frequent being adenovirus genes E1A and E1B that have been used to immortalize various human cells like HEK293, PerC.6 and CAP cells and the human papilloma virus E6 and E7 genes in the HeLa cell line). When tumorigenic cell lines are used as substrate, the purification process must be capable of reducing as much as possible residual DNA, and a specific testing to demonstrate absence or fragmentation of the oncogenic genes is necessary [23,24]. Particularly, DNA cleavage to fragments shorter than 200 bp is considered as a reasonable risk mitigation for oncogenicity. As AAV capsids can contain host cell DNA, this target is likely to be unattainable. In this case, specific studies should be conducted to demonstrate the product safety. Although fragments of helper virus DNA and plasmids are also packaged in AAV capsids and have to be controlled as any other impurity, they are not subject to such stringent specifications as long as they do not contain oncogenic sequences.
Host cell proteins (HCPs) in sufficient quantities may be associated with immunogenicity, inflammation, or anaphylactic shock. When human cells are used, the HCPs are expected to be poorly immunogenic unlike non-human material (helper viruses like HSV, Adenovirus, Baculovirus and non-human cell lines like BHK21 or insect cells). In any case, considering the massive doses of AAV to be administered to patients, the risk associated with HCPs and/or viral proteins is not negligible. DSP has to reduce residual proteins as much as possible.
The impact of empty capsids is not defined clearly yet. In the first Hemophilia B clinical trials, a cell-mediated immune response against the AAV capsid antigens was detected and correlated with the loss of transgene expression [25]. This observation suggests that removing empty capsids would help reducing the anti-AAV immune response. However, this idea is controversial since empty particles are also suspected to act as a decoy for low levels of neutralizing antibodies [26]. As there is no consensus on empty capsids, the profile of clinical-grade AAV vectors is variable with some products containing up to 90% empty capsids [16] while some downstream processes include a full capsid enrichment [27].
Other impurities may be introduced during the process and have to be eliminated and controlled in the drug substance, especially if they present a risk of toxicity for the patients. The nature of these impurities depends on the raw materials entering the process, but frequent examples are Bovine Serum Albumin (BSA) when the culture medium contains fetal calf serum, nuclease and detergent that are used to reduce DNA and to disrupt the cells, respectively.
Finally, when the upstream process relies on a viral expression system (like HSV, Adenovirus or Baculovirus), it is mandatory to demonstrate that the final product is free of infectious viruses [12].
With all these points in mind, the downstream process engineers have to design specific steps and deploy several kinds of technologies to reach the highest level of purity while avoiding damages or loss of the vector during processing.
Downstream Processing at industrial scale
The historical methods to purify AAV involve centrifugation and cesium chloride density isopycnic gradients [28]. As an alternative to the heavy metal toxicity of cesium, iodixanol can be used [29]. Isopycnic gradients have the advantage to separate full capsids from the empty particles but are not convenient for commercial GMP manufacturing due to their limited scalability. Futhermore, depending on the protocol and the number of ultracentrifugation runs, density gradients alone may not suffice to remove HCPs efficiently [30].
At industrial scale, other technologies are available for scalable and reproducible DSP. Several methods are combined in a succession of key steps that are common to many biological processes. Typical flow charts of AAV purification are given as an example in Figure 1.
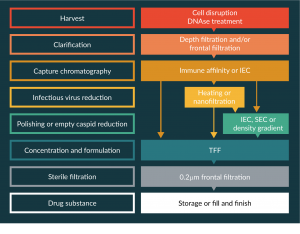
Figure 1: Examples of process flow charts for AAV purification at industrial scales.
Cell lysis & elimination of cell debris
AAV is not released very efficiently from the cells, although major differences have been observed between serotypes [31]. When harvesting the culture, a cell disruption method is usually applied to recover the vectors entrapped in the cells. It can be either a detergent-based chemical lysis or a mechanical method such as freeze-thaw cycles, homogenization or osmotic shock [27]. In a GMP environment, mechanical lysis implies the use of devices requiring the fastidious process of cleaning validation, therefore the use of detergent is widely used.
The viruses used as expression systems can have a lytic cycle in the host cells, which may help to release AAV capsids. However, as the cell death may be delayed compared to the AAV productivity peak, lysis method can be beneficial to standardize the harvest day and the bulk homogeneity.
After lysis, the harvest becomes a highly turbid suspension of DNA, proteins and large cell debris. The first step to remove most of this material – at least the macromolecular complexes – is clarification. Centrifugation is a standard procedure at small scale but less frequently used in manufacturing where only a continuous centrifuge system is applicable. Therefore, the common clarification system in the industry is filtration. Many types of filters are available in the market: membrane filters of various porosities or depth filters. Depth filtration allows reducing the filter surface area, which is more convenient for industrial scale [32].
The optimal filters must be defined for each product because both the upstream process characteristics (particularly the cell mass at harvest) and the AAV serotype (which may adsorb to the filter membrane) can have a major impact on the clarification performance. The endpoint is to obtain a low turbidity in the clarified harvest to avoid clogging at the next chromatography step.
DNA reduction using nucleases
Nuclease treatment has become a standard element in biomanufacturing since the commercialization of Benzonase. Although the cost of recombinant enzymes is elevated for large-scale processes, this step offers several benefits for product purification. Firstly, it solves a technical issue associated with cell DNA: after cell disruption, genomic DNA forms aggregates and macromolecular complexes that tend to clog the filters and column chromatography media, and increase viscosity that extends the process duration [33]. As nucleases cleave genomic DNA into small fragments, the crude cell lysate is more homogenous and shows higher filterability. Secondly, nucleases can break host cell DNA into small fragments, thus decreasing the risk of carrying over full-length oncogenes in the final product.
Nuclease treatment can be performed at various steps of the process. To limit the complexes and viscosity induced by genomic DNA, it is preferable to add the enzyme directly in the culture concomitantly with cell breakage. However, besides being costly, this step may not be compatible with the optimal conditions for enzyme activity. For instance, in the Baculovirus process, the Sf9 culture medium has a pH below 6. Optimal pH for Benzonase being 6-10, the enzyme will be poorly active in this condition [33]. Additionally, NaCl concentration of the culture medium may alter the enzyme efficacy depending on the nuclease type [33,34].
Alternatively, nucleases can be added after clarification or after chromatography but in this case, the benefit of the enzyme is moderate since most of the DNA is eliminated by the previous steps.
Process impurities reduction using Capture Chromatography
The most effective technology to separate biological products from their impurities is capture chromatography. The purpose is to bind the AAV vector to a matrix in a specific manner to wash away the HCPs, helper virus proteins, DNA and other impurities like detergent and benzonase through a dynamic flow.
As AAV capsids possess positive and negative charges on their surface, they can be retained by ion exchange chromatography (IEC). Depending on the isoelectric point of the capsids and the pH, the global charges of the capsid can be negative or positive, allowing the use of either anion-exchange chromatography or cation-exchange chromatography. A variety of IEC resins is commercially available and many of them have been reported to be effective for AAV purification. Among the resins frequently used, strong anion exchanger based on quaternary amine (like POROS HQ, ThermoFisher) or strong cation exchanger based on sulphopropyl (like SP Sepharose, GE Healthcare) have been described for the purification of diverse AAV serotypes (AAV1, AAV2, AAV4, 5, 6, 8, 9) [7,35–39].
Besides IEC based on functionalized resins that need to be packed in columns, membrane chromatography can be used. They rely on similar ligand chemistry (for instance, quaternary amine in Mustang Q and sulfonic acid in Mustang S, Pall) while the ligands are bound to polyethersulfone (PES) membrane [40,41]. Although this kind of device is theoretically recyclable, it becomes more convenient than column-packed resins when used as a single-use plug-in device.
Although IEC is efficient to capture AAV vectors, it cannot be highly selective since many other impurities may display an isoelectric point close to AAV’s, resulting in co-purification of undesired proteins or DNA. Consequently, a capture IEC typically requires an additional chromatography step using another matrix and different pH and salt conditions. As an alternative, immune affinity (IA) chromatography is a highly specific capture step. Several references have been developed specifically for AAV vectors. All the commercial resins rely on the VHH fragment of a camelid antibody specific for a given AAV serotype (Table 2). The VHH fragment is a 12 to 14 kD polypeptide manufactured in yeast and covalently bound to a resin matrix: sepharose or Polystyrene-Divinylbenzene (POROS) beads.
| Table 2: AAV-specific immunoaffinity resins currently available on the market. | ||
|---|---|---|
| Immune affinity resin | Supplier | AAV serotype specificity |
| AVB Sepharose HP | GE Healthcare | AAV1, 2, 3, 5, 6, 8, AAVrh10 |
| POROS™ CaptureSelect™ AAV8 | Thermofisher | AAV8 |
| POROS™ CaptureSelect™ AAV9 | Thermofisher | AAV9 |
| POROS™ CaptureSelect™ AAVX | Thermofisher | AAV1, 2, 3, 4, 5, 6, 7, 8, 9, AAVrh10 and synthetic serotypes |
AVB Sepharose was the first IA resin commercialized for AAV purification. Initially developed for AAV1, the ligand also displays affinity for AAV2, 3, 5, 6 and AAVrh10. The resin can also be used to purify AAV8, however, the vector recovery is usually low (below 50%) due to low affinity for this serotype resulting from amino acid variation in the target epitope [42].
As the demand for AAV8 and AAV9 serotypes increased, VHH libraries were screened to develop the highly specific resins POROS™ CaptureSelect™ AAV8 and POROS™ CaptureSelect™ AAV9. Recently, ThermoFisher commercialized POROS™ CaptureSelect™ AAVX. This resin shows a very high affinity for every serotype tested so far suggesting the existence of a highly conserved epitope on AAV capsids. Of note, POROS resins are designed with a high density of ligands, which results in an enormous binding capacity up to 1E14 vg/mL of resin [43]. Considering that most of the upstream processes can deliver 1E13 to 1E14 vg/L, one milliliter of POROS beads should suffice to purify 1 to 10L of a culture harvest.
Due to the specificity of the antibody, IA resins bind very few contaminants. In addition, due to high avidity, the wash conditions can be very stringent to desorb potential impurities. Therefore, unlike IEC, one single IA chromatography step can deliver a highly purified AAV.
Polishing
In many bioprocesses, polishing aims at further decreasing impurity levels, especially HCPs. Polishing might not be required when IA is used as the capture step. However, it can be useful to improve the product quality, particularly to eliminate the chromatography ligand (VHH) that potentially leaches from IA resin during elution.
Polishing is usually based on IEC or size exclusion chromatography (SEC) [39]. For this purpose, IEC can be operated either in a positive mode (capture chromatography) or in a negative mode (flow through). SEC has been successfully used for AAV vectors [16] but usually it is not the most popular option in the industry because its scalability is more complex than IEC.
It is important to keep in mind that no step of a downstream process can give 100% vector recovery. Therefore, before implementing polishing, it is recommended to determine the balance between the benefit for purity and the process yield.
Reducing empty capsids
Production of AAV vectors is inevitably accompanied by empty capsids. Whether it has a biological importance or not is not yet clear [26,44]. However, for some clinical trials, the manufacturers have implemented a specific step to decrease their quantity [15].
The historical method is based on ultracentrifugation through a density gradient [28]. The gradient can be made of cesium chloride or iodixanol [29,30]. Even though this procedure has been used for manufacturing of GMP INDs, it is not convenient to treat large volumes at industrial scale and is difficult to standardize in a GMP environment because the genome-containing capsids are usually collected from the gradient using a needle through the tube wall. The precision of the gesture relies on operator’s training and on visual subjectivity to locate the material in the gradient. In case of inappropriate puncture of the tube, the product may be poorly recovered or completely lost.
Alternatively, the use of continuous ultracentrifugation has been reported to enrich the amount of full capsids [45]. This process has been used for decades in the vaccine industry, for instance to purify influenza virus antigens [46]. It is a very attractive approach for large-scale separation of AAV capsids. However, it requires specific equipment and a high level of know-how, which is usually limited to large pharmaceutical companies. Of note, yield and efficiency of this method to segregate full capsids from empty capsids were not communicated.
The most promising option to implement full capsids enrichment at industrial scale is based on an ion exchange chromatography step. In 2007, Qu et al. described that AAV2 empty capsids displayed slight differences of charge properties compared to their genome-containing counterparts, which resulted in a possible separation using the anion exchange matrix Q Sepharose [36]. Since then, this has been confirmed in other serotypes and on other matrices, like the monolith columns CIM-Q [47]. The complexity of this approach is that the charge differences between empty and full capsids is narrow, so the differential elution of the viral particles can be achieved only with a very slow and flat linear gradient of sodium chloride. When successful, it results in the elution of 2 peaks very close to each other. The proximity of the peaks renders the operation difficult at large scale. Usually, in GMP conditions, standardized procedures involving established volumes and accurate timing are preferred to avoid operator-induced variations and to guarantee batch-to-batch consistency.
In summary, even though three technologies allow separating full from empty capsids in AAV preparations, none of them is easily transposable to large-scale manufacture. Further development work is needed to optimize and to standardize these methods.
Viral risk management
In every biological process, viral risk is a major concern: in case of adventitious virus introduction in USP, the virus may propagate in the producer cells and may contaminate the final product. Viral vector manufacture is particularly at risk since DSP is designed to preserve the therapeutic virus and thus will offer limited capability to eliminate adventitious agents.
For this reason, as defined in regulatory guidelines [11], viral safety is guaranteed by testing and qualifying the raw materials. Particularly, the biological starting materials like the Master Cell bank and the Master Virus Seed have to be tested for adventitious viruses. Any material of animal origin (like bovine serum), has to be controlled too and preferably should be irradiated. On top of that, since there is a risk of introducing adventitious agents by manual operations during the process, it is also mandatory to control each harvest before downstream processing.
Eventually, the purification process must be validated for viral clearance, meaning for its capacity to reduce, inactivate or eliminate model viruses that are inoculated on purpose at key steps in a scale down system [11]. Viral clearance validation is mandatory for Phase III clinical trials and commercial phase. However, in the case of viral expression systems like Baculovirus, HSV or Adenovirus, massive amounts of infectious virus are present in harvest. Hence, even for phase I clinical trials, the infectious viruses have to be eliminated by an appropriate method and the final product must be demonstrated to be free of the corresponding virus [12].
To eliminate infectious viruses, several options have been described for AAV manufacturing processes:
The use of detergent to release AAV from the cells is also very effective in disrupting enveloped viruses like baculovirus and HSV but has no or limited effect on the non-enveloped adenovirus.
Heating can be used to inactivate adenovirus (AdV), since AAV is slightly more resistant than AdV to temperature above 50°C [48]. This approach appears challenging to scale up, since any variation of the temperature or exposure time may result in either AAV inactivation or insufficient killing of AdV.
For clearance validation, it is usually necessary to include several virus reduction steps in the process. The clearance capacity of a process is defined as sum of the logarithm of the reductions at each step [11] and must exceed the initial infectious titer of the preparation. For instance, if the harvest of BEVS-derived AAV batch contains 1E8 PFU/mL of Baculovirus, the purification process must reduce the infectivity of at least 8 log10 to claim a risk of residual infectivity <1PFU/mL. If the volume of AAV to be administered to the patient is 100 mL, then the process clearance capacity must be higher than 10 log10 to guarantee less than 1PFU in 100 mL. To achieve a high level of viral clearance, uniQure developed and patented nanofiltration [49]. Nanofilters are designed to remove viruses larger than 50 nm. AAV being smaller than the cutoff of the filter, it can be separated from Baculovirus.
Therefore, with a combination of detergent, nanofiltration and chromatography, efficient viral reduction is achievable for large enveloped viruses. As an example, Ye et al. were able to attain a clearance higher than 14 log10 for HSV-1 in three steps (Triton X100, CIM Q and AVB Chromatography) from the Herpesvirus expression system [50].
This section is also a good opportunity to remind that Sf9 cells have been reported to contain nucleic acid sequences from an insect Rhabdovirus [51]. Even though this virus does not seem to be infectious depending on the Sf9 cells origin [52], it may be a safety concern so Baculovirus-derived AAV vectors should be tested for it. Alternatively, other insect cell lines free of Rhabdovirus could be used in the upstream process.
Concentration and diafiltration
The final step of downstream process is the concentration of the vector to the desired titer and the formulation in a buffer appropriate for stability of the product and patient administration.
The standard method is tangential flow filtration (TFF) which can be carried out on hollow fibers or on filter cassettes [7,39]. As a non-enveloped virus, AAV is highly resistant to shear stress. This offers flexibility for the operating conditions in terms of flow rate, transmembrane pressure and process duration.
Due to the small vector size, the membrane cutoff cannot exceed 100 kD or 300 kD [39]. Such a tight porosity does not allow many impurities to pass through. As a consequence, TFF cannot be considered as a purification step. Thus, the vector has to be sufficiently purified prior to TFF; otherwise, most of the remaining impurities will be concentrated as well.
Nevertheless, using a 300 kD membrane, TFF can theoretically eliminate the leaching VHH ligand of IA resins (12 kD) reinforcing the fact that polishing step may be dispensable when IA chromatography is used as the capture step.
The main technical risk for TFF is the possible aggregation of AAV vectors at high concentration. This has been shown for AAV2 serotype [53]. Only the buffer composition, particularly high salt concentrations, can alter the aggregation point. Therefore, purity, buffer, serotype and target titer are the four major interacting critical parameters in TFF development.
Global process yield
The details of downstream processes used to manufacture clinical grade AAV vectors are rarely communicated, especially the percentage of product recovery. This being said, every manufacturing expert will agree that a purification step can hardly give a yield higher than 90%. Considering a 5-step process (clarification, capture chromatography, polishing, TFF and sterile filtration) with 90% recovery per step, the theoretical overall yield can be no more than 60%. The value drops to 32% for a more realistic recovery per step of 80%. These yields are consistent with the ones that were obtained experimentally in our laboratory for AAV8 and AAV9 serotypes (40% and 60%, respectively) [54,55].
Translational Insights
After several clinical successes, AAV vectors are entering the industrial phase to support phase III trials and commercialization. Improving productivity and cost effectiveness remain as major challenges, as treatment costs will be a serious impediment for the future of gene therapy.
Regarding downstream processing, most of the technologies are already available to support large scale production in GMP environment. The recent development of high-capacity immune affinity resins offers a potent and scalable solution for efficient chromatography. This being said, manufacturers should keep in mind that each new AAV product can display new characteristics, for instance with regard to titers, empty to full capsid ratio and adsorption or aggregation related to the serotype. Therefore, developing a generic DSP to purify every AAV vector is not necessarily attainable or at least might not be optimal in every case.
The major uncertainty in the area of DSP is related to product purity. Considering the increasing number of clinical trials with AAV gene therapy, and the corresponding amount of safety data that are generated continuously, one should expect a harmonization of the quality requirements for AAV vectors in the near future. Particularly, these trials should provide more information on the biological role of empty particles and on the harmlessness of encapsidated oncogenic sequences from host cell DNA. In any case, it is reasonable to think that the medicine agencies will ultimately set up some rules and specifications for these attributes. Thus, the process developers are encouraged to continue creating and ameliorating technologies to eliminate and control more stringently impurities in a standardized and reproducible manner.
Financial & competing interests disclosure
The author has no relevant financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock options or ownership, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
1. Kishi Y, Kami M, Miyakoshi S et al. Early immune reaction after reduced-intensity cord-blood transplantation for adult patients. Transplantation 2005; 80(1), 34–40.
CrossRef
2. Hauswirth WW, Aleman TS, Kaushal S et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther. 2008; 19(10), 979–90.
CrossRef
3. Nathwani AC, Tuddenham EG, Rangarajan S et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011 Dec 22;365(25):2357-65.
CrossRef
4. Avexis. AveXis Announces New England Journal of Medicine Publication of Phase 1 Data of AVXS-101 Gene Replacement Therapy in Spinal Muscular Atrophy Type 1. Press Release November 1, 2017.
5. Audentes. Audentes Announces Positive Interim Data from First Dose Cohort of ASPIRO, a Phase 1/2 Clinical Trial of AT132 in Patients With X-Linked Myotubular Myopathy. Press Release January 4, 2018.
6. European Medicines Agency website. Glybera, alipogene tiparvovec. uniQure biopharma B.V. Marketing authorization 25 October 2012. Website
7. Food and Drug Administration website. Luxturna, voretigene neparvovec-rzyl. Spark Therapeu-tics Inc. Approval letter December 19, 2017. Website
8. Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clini-cal trials. Mol. Ther. Methods Clin Dev. 2016; 3, 16002.
CrossRef
9. Merten OW. AAV vector production: state of the art developments and remaining challenges. Cell & Gene Therapy Insights. 2016; 2(5), 521–51.
CrossRef
10. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-Associated Virus (AAV) as a Vec-tor for Gene Therapy. BioDrugs. 2017 31(4), 317–34.
CrossRef
11. Dismuke DJ & Kotin RM. Obstacles for rAAV clinical trials: a question of vector supply and demand or know-how. Cell Gene Therapy Insights. 2017; 3(9), 755–68.
CrossRef
12. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Viral safety evaluation of biotechnology products derived from cell lines of human origin. Q5A(R1). Website
13. European Medicines Agency website. Ph.Eur. monograph 5.14 Gene transfer medicinal prod-ucts for human use. 01/2010. Website
14. Mietzsch M, Grasse S, Zurawski C et al. OneBac: platform for scalable and high-titer produc-tion of adeno-associated virus serotype 1-12 vectors for gene therapy. Hum. Gene Ther. 2014; 25(3), 212–22.
CrossRef
15. Mietzsch M, Hering H, Hammer EM, Agbandje-McKenna M, Zolotukhin S, Heilbronn R. One-Bac 2.0: Sf9 Cell Lines for Production of AAV1, AAV2, and AAV8 Vectors with Minimal Encapsi-dation of Foreign DNA. Hum. Gene Ther. Methods 2017; 28(1), 15–22.
CrossRef
16. Grieger JC, Soltys SM, Samulski RJ. Production of Recombinant Adeno-associated Virus Vectors Using Suspension HEK293 Cells and Continuous Harvest of Vector from the Culture Me-dia for GMP FIX and FLT1 Clinical Vector. Mol. Ther. 2016; 24(2), 287–97.
CrossRef
17. Allay JA, Sleep S, Long S et al. Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum. Gene Ther. 2011; 22(5), 595–604
CrossRef
18. Lecomte E, Tournaire B, Cogné B et al. Advanced Characterization of DNA Molecules in rAAV Vector Preparations by Single-stranded Virus Next-generation Sequencing. Mol. Ther. Nu-cleic Acids. 2015; 4, e260.
CrossRef
19. Hauck B, Murphy SL, Smith PH et al. Undetectable transcription of cap in a clinical AAV vec-tor: implications for preformed capsid in immune responses. Mol. Ther. 2009; 17(1), 144–52.
CrossRef
20. Wright JF. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Charac-terization and Risk Assessment. Biomedicines 2014; 2, 80–97.
CrossRef
21. Penaud-Budloo M, Lecomte E, Guy-Duché A et al. Accurate Identification and Quantification of DNA Species by Next-Generation Sequencing in Adeno-Associated Viral Vectors Produced in Insect Cells. Hum. Gene Ther. Methods 2017; 28(3), 148–62.
CrossRef
22. Loring HS, ElMallah MK, Flotte TR. Development of rAAV2-CFTR: History of the First rAAV Vector Product to be Used in Humans. Hum. Gene Ther. Methods 2016; 27(2), 49–58.
CrossRef
23. Food and Drug Administration website. Cellular and Gene Therapy Guidances. Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). 4/2008. Website
24. Food and Drug Administration website. Guidance for Industry. Characterization and Qualifica-tion of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications. 2010. Website
25. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Derivation and characteri-zation of cell substrates used for production of biotechnological/biological products. Q5D. 1997. Website
26. Mingozzi F, Maus MV, Hui DJ et al. CD8(+) T-cell responses to adeno-associated virus cap-sid in humans. Nat. Med. 2007; 13(4), 419–22.
CrossRef
27. Mingozzi F, Anguela XM, Pavani G et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013; 5(194), 194.
CrossRef
28. Burova E, Ioffe E. Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: methods and implications. Gene Ther. 2005; 12 Suppl 1, S5–17.
CrossRef
29. Rose JA, Hoggan MD, Shatkin AJ. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proc. Natl Acad. Sci. USA 1966; 56(1), 86–92.
CrossRef
30. Zolotukhin S, Byrne BJ, Mason E et al. Recombinant adeno-associated virus purification us-ing novel methods improves infectious titer and yield. Gene Ther. 1999; 6(6), 973–85.
CrossRef
31. Strobel B, Miller FD, Rist W, Lamla T. Comparative Analysis of Cesium Chloride- and Iodixa-nol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applica-tions. Hum. Gene Ther. Methods 2015; 26(4),147–57.
CrossRef
32. Vandenberghe LH, Xiao R, Lock M, Lin J, Korn M, Wilson JM. Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 2010; 21(10), 1251–7.
CrossRef
33. Buyel JF, Gruchow HM, Fischer R. Depth Filters Containing Diatomite Achieve More Efficient Particle Retention than Filters Solely Containing Cellulose Fibers. Front. Plant Sci. 2015; 6, 1134.
CrossRef
34. Merck Millipore Website. Benzonase® Endonuclease Brochure. Website
35. ArcticZymes® Website. Salt Active Nuclease. Website
36. Urabe M, Xin KQ, Obara Y et al. Removal of empty capsids from type 1 adeno-associated vi-rus vector stocks by anion-exchange chromatography potentiates transgene expression. Mol. Ther. 2006; 13(4), 823–8.
CrossRef
37. Qu G, Bahr-Davidson J, Prado J et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J. Virol. Methods 2007; 140(1–2), 183–92.
CrossRef
38. Kaludov, Handelman B, Chiorini JA. Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Hum. Gene Ther. 2002; 13(10), 1235–43.
CrossRef
39. Brument N, Morenweiser R, Blouin V et al. A versatile and scalable two-step ion-exchange chromatography process for the purification of recombinant adeno-associated virus serotypes-2 and -5. Mol. Ther. 2002; 6(5), 678–86.
CrossRef
40. Qu W, Wang M, Wu Y, Xu R. Scalable downstream strategies for purification of recombinant adeno- associated virus vectors in light of the properties. Curr. Pharm. Biotechnol. 2015; 16(8), 684–95.
CrossRef8
41. Davidoff AM, Ng CY, Sleep S et al. Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J. Virol. Methods. 2004; 121(2), 209–15.
CrossRef
42. Okada T, Nonaka-Sarukawa M, Uchibori R et al. Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum. Gene Ther. 2009; 20(9, 1013–21.
43. Wang Q, Lock M, Prongay AJ, Alvira MR, Petkov B, Wilson JM1. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol. Ther. Methods Clin. Dev. 20154; 2, 15040.
44. ThermoFisher website. Data Sheet: POROS CaptureSelect AAV Affinity Resins. Website
45. Gao K, Li M, Zhong L et al. Empty Virions In AAV8 Vector Preparations Reduce Transduction Efficiency And May Cause Total Viral Particle Dose-Limiting Side-Effects. Mol. Ther. Methods Clin. Dev. 2014; 1(9), 20139.
CrossRef
46. Bendik M. AAV Vector Quantification Strategy for Clinical Trials. Presented at ISBiotech, Washington DC, USA, March 6, 2017.
47. Reimer CB, Baker RS, Van Frank RM, Newlin TE, Cline GB, Anderson NG. Purification of large quantities of influenza virus by density gradient centrifugation. J. Virol. 1967; 1(6), 1207–16.
48. Lock M, Alvira MR, Wilson JM. Analysis of particle content of recombinant adeno-associated virus serotype 8 vectors by ion-exchange chromatography. Hum. Gene Ther. Methods 2012;23(1), 56–64.
CrossRef
49. Thorne B. Towards Industrial Scale Manufacturing of MYDICAR®: AAV1/SERCA2a for Heart Failure. Presented at ISBiotech, Washington DC, USA, March 11, 2015.
50. Hermens WTJMC and Smith JP. Removal of contaminating viruses from AAV preparations. WO2013036118 A1
51. Ye GJ, Scotti MM, Thomas DL, Wang L, Knop DR, Chulay JD. Herpes simplex virus clear-ance during purification of a recombinant adeno-associated virus serotype 1 vector. Hum. Gene Ther. Clin. Dev. 2014; 25(4), 212–7.
CrossRef
52. Ma H, Galvin TA, Glasner DR, Shaheduzzaman S, Khan AS. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J. Virol. 2014; 88(12), 657–85.
CrossRef
53. Hashimoto Y, Macri D, Srivastava I et al. Complete study demonstrating the absence of rhabdovirus in a distinct Sf9 cell line. PLoS One 2017; 12(4)
CrossRef
54. Wright JF, Le T, Prado J et al. Identification of factors that contribute to recombinant AAV2 particle aggregation and methods to prevent its occurrence during vector purification and formula-tion. Mol. Ther. 2005; 12(1), 171–8.
CrossRef
55. Hebben M. A New Downstream Process for Large Scale Manufacturing of AAV9 Vectors. Presented at ESGCT, The Hague, October 25, 2014.
56. Hebben M. Large Scale Manufacture of AAV Vector: Scale up and Transfer of AAV Produc-tion Process for Gene Therapy of Crigler Najjar Syndrome. Presented at ISBiotech, Washington DC, March 8, 2017.
Affiliation
Matthias Hebben
Généthon, 1 bis, rue de l’Internationale, 91000 Evry, France.
mhebben@genethon.fr

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License</