
Breakthrough Therapy Designation & Regenerative Medicine Advanced Therapy Designation Programs in Cellular & Gene Therapies
Cell Gene Therapy Insights 2018; 4(6), 563-571.
10.18609/cgti.2018.046
A key responsibility of the US Food and Drug Administration (FDA) is to facilitate availability of innovative, safe, and effective treatments to patients. The FDA utilizes several expedited programs to speed development of exceptionally promising therapies for serious or life-threatening conditions. The newest expedited programs are the Breakthrough Therapy Designation (BTD) and the Regenerative Medicine Advanced Therapy (RMAT) designation. The marketing approvals in 2017 of three gene therapy products with BTD represent the success of the BTD program. The 21st Century Cures Act (Cures Act) defines regenerative medicine therapy and creates the RMAT designation, bringing hope for earlier availability of new regenerative medicine therapies, including cellular therapies and certain gene therapies. This article provides an overview of the BTD and RMAT designation programs, and a summary of the experience with these programs in FDA’s Office of Tissues and Advanced Therapies (OTAT) in FDA’s Center for Biologics Evaluation and Research (CBER).
Submitted: May 5th 2018 Published: 7th July 2018
The US Food and Drug Administration (FDA) is responsible for advancing public health by helping to speed innovations that make medicines more effective and safer. Several times over the past three decades, the US Congress has enacted legislation to amend the Federal Food, Drug, and Cosmetic (FD&C) Act to establish programs that expedite the development and review of products for serious or life-threatening conditions. To date, FDA has implemented five such programs:
- Priority Review (1992)
- Accelerated Approval (1992)
- Fast Track designation (1997)
- Breakthrough Therapy designation (BTD) (2012)
- Regenerative Medicine Advanced Therapy (RMAT) designation (2016)
Regulation of cellular and gene therapy products is the responsibility of the Office of Tissues and Advanced Therapies (OTAT; formerly the Office of Cellular, Tissue and Gene Therapies (OCTGT)), in the Center for Biologics Evaluation and Research, FDA (OTAT / CBER / FDA). Requests for BTD and RMAT designation for these products are reviewed by OTAT in the context of an investigational new drug application (IND) submitted by industry or academic sponsors.
Here, we describe the two most recent programs, BTD and RMAT designation, and summarize OTAT’s experience with these programs to date.
BREAKTHROUGH THERAPY DESIGNATION AND REGENERATIVE MEDICINE ADVANCED THERAPY DESIGNATION
[1,2]
Established under the FDA Safety and Innovation Act, passed on July 9, 2012, BTD is intended to expedite the development and review of novel drugs and biological products for serious conditions. To qualify, a product must be intended to treat a serious or life-threatening disease or condition, and the preliminary clinical evidence indicates that the product may demonstrate substantial improvement over available therapies on one or more clinically significant endpoints. Sponsors whose products are granted this designation are eligible to receive intensive guidance from FDA on an efficient product development program, beginning as early as Phase 1. In addition, sponsors receive FDA’s organizational commitment to involve senior management in facilitating development of the product.
On December 13, 2016, President Obama signed into law the 21st Century Cures Act (Cures Act). The Cures Act defines “regenerative medicine therapy” as including cell therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products, except for those regulated solely under section 361 of the Public Health Service Act (42 USC 264) and Title 21 of the Code of Federal Regulations Part 1271. Additionally, as specified in the FDA Draft Guidance for Industry: Expedited Programs for Regenerative Medicine Therapies for Serious Conditions (2017), FDA interprets the definition of a regenerative medicine therapy to include certain gene therapies.
Section 3033 of the 21st Century Cures Act states that a drug is eligible for RMAT designation if:
- It meets the definition of a regenerative medicine therapy;
- It is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and
- Preliminary clinical evidence indicates that the regenerative medicine therapy has the potential to address unmet medical needs for such disease or condition.
Benefits of RMAT designation include all those of BTD, as well as the opportunity for early interactions with FDA to discuss potential surrogate or intermediate endpoints to support accelerated approval for marketing.
Table 1 compares the key features of BTD and RMAT designation [1].
| Table 1 Comparison of key features of the Breakthrough Therapy designation and Regenerative Medicine Advanced Therapy designation programs. | |||
|---|---|---|---|
| Statute | Section 506(a) of the FD&C Act, as added by section 902 of the Food and Drug Administration Safety and Innovation Act (FDASIA) (2012) | Section 506(g) of the FD&C Act, as added by section 3033 of the 21st Century Cures Act (2016) | |
| Qualifying criteria | A drug (including biological product), AND the drug is intended to treat a serious condition, AND preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies | A drug is a regenerative medicine therapy, AND the drug is intended to treat, modify, reverse, or cure a serious condition, AND preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition | |
| Benefits | Actions by FDA to expedite development and review Rolling review* Intensive guidance from FDA on efficient drug development, beginning as early as Phase 1 Organizational commitment involving FDA senior managers | All the benefits of BTD Early interactions to discuss any potential surrogate or intermediate endpoints; Statute describes ways to potentially support accelerated approval and satisfy post-approval requirements. | |
| When to submit | With the original submission of an Investigational New Drug application (IND) or after the IND is active, and, ideally, no later than the end-of-phase 2 meeting | ||
| FDA response | Within 60 calendar days after receipt of request | ||
| Designation rescission | Designation may be rescinded later in product development if the product no longer meets the designation-specific qualifying criteria. | ||
| *Rolling review means that an applicant (e.g., a pharmaceutical company) can submit completed section(s) of its marketing application, such as a Biologics License Application (BLA) for review by FDA, rather than waiting until every section of the BLA is completed before submitting the entire application for review. | |||
OTAT EXPERIENCE WITH THE BREAKTHROUGH THERAPY DESIGNATION PROGRAMME
Since the launch of the program in July 2012, OTAT has received and reviewed 93 BTD requests from sponsors of active INDs. Of these, 28 requests (30%) were granted, 62 (67%) were denied, and three (3%) were voluntarily withdrawn by the sponsor prior to FDA’s decision (Figure 1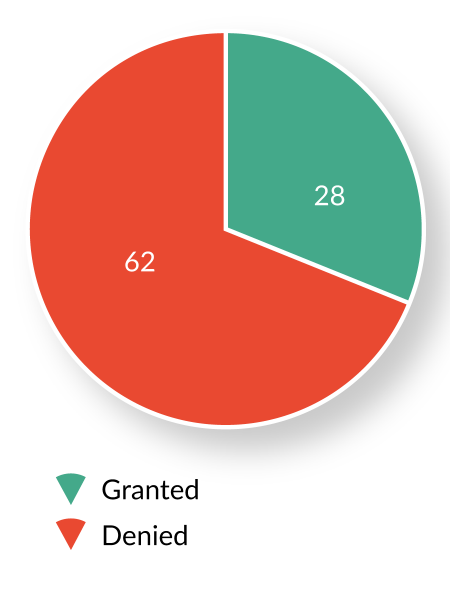
| Table 2 BTD by product type (excluding withdrawn and pending requests). | |
|---|---|
| Product | Granted/requests (%) |
| Cellular Therapy | 3/26 (12%) |
| Gene Therapy | 21/45 (47%) |
| Others | 4/19 (21%) |
| Table 3 BTD by product type (excluding withdrawn and pending requests). | |
|---|---|
| Indications | Granted/requests (%) |
| Oncololgy (solid tumor) | 6/34 (18%) |
| Hematology (malignant and benign) | 16/25 (64%) |
| Others | 6/31 (19%) |
Both Table 3 and Figures 2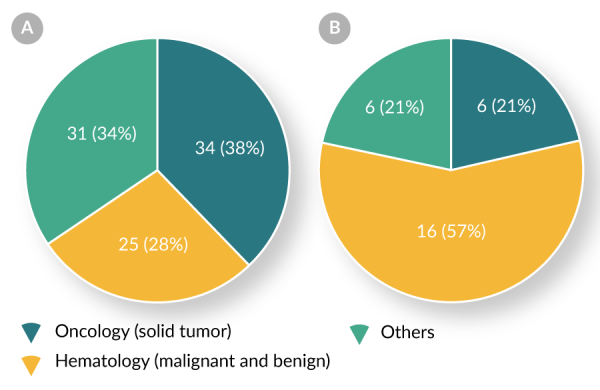
The most frequent reasons for denial of BTD have been:
- Clinical evidence was too preliminary to be considered reliable (e.g., an early-phase, /open-label study with a small sample size and without suitable natural history data to serve as a historical control);
- Improvement over available therapy did not appear substantial; and
- The original product underwent major modifications during clinical development, such that the clinical evidence generated using the original product could not be used to support BTD for the modified product.
Marketing approval of products granted breakthrough therapy designation
In 2017, five years after implementation of the BTD program, OTAT granted marketing approval to three products that had received BTD. Two (tisagenlecleucel and axicabtagene ciloleucel) are genetically-modified chimeric antigen receptor (CAR) T cell products, and the third (voretigene neparvovec-rzyl) is an adeno-associated virus (AAV) vector-based gene therapy product.
Tisagenlecleucel is the first cell-based gene therapy approved in the United States. It is composed of autologous human T cells genetically modified with a lentiviral vector encoding a CAR. The receptor specifically recognizes CD19, an antigen expressed on the surface of most B-cell malignancies, as well as on normal B lymphocytes. Tisagenlecleucel is indicated for the treatment of patients 3 to 25 years of age with relapsed or refractory B-cell acute lymphoblastic leukemia. Tisagenlecleucel received BTD in April 2016, and marketing approval on August 30, 2017.
Axicabtagene ciloleucel comprises autologous human T cells transduced with a retroviral vector encoding a CAR directed against human CD19. Axicabtagene ciloleucel is indicated for treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. Axicabtagene ciloleucel received BTD in December 2015, and marketing approval on October 18, 2017.
Voretigene neparvovec-rzyl is the first gene therapy approved in the US targeting a genetic disease due to single gene mutations. Voretigene neparvovec-rzyl consists of a recombinant AAV serotype 2 vector expressing the RPE65 gene, encoding human retinal pigment epithelium 65 kDa protein. The product is indicated for treatment of adult and pediatric patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Voretigene neparvovec-rzyl received BTD in October 2014 and marketing approval on December 19, 2017.
OTAT Experience with Regenerative Medicine Advanced Therapy Designation
Since establishment of the RMAT designation program in December 2016, OTAT has received and reviewed 51 RMAT designation requests from sponsors of active INDs with ongoing development programs. Of these, 19 (37%) requests were granted, 29 (57%) were denied, and 3 (6%) were voluntarily withdrawn by the sponsors prior to an OTAT decision (Figure 3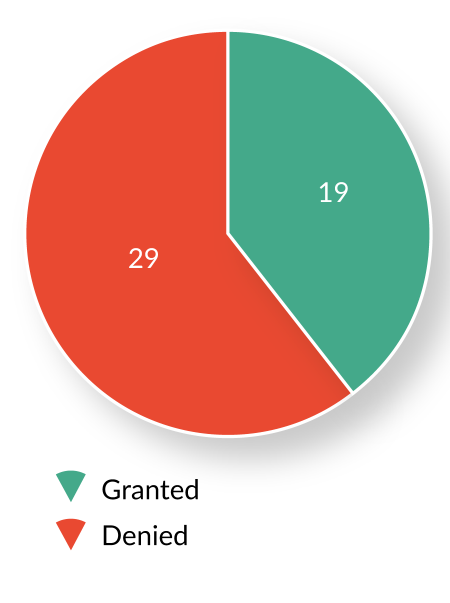
| Table 4 RMAT designation by product type (excluding withdrawn and pending reque sts). | |
|---|---|
| Products | Granted/requests (%) |
| Allogeneic cellular therapy | 11/23 (48%) |
| Autologous cellular therapy | 2/11 (18%) |
| Gene therapy | 5/8 (63%) |
| Others | 1/6 (17%) |
While the number of BTD requests and granted BTDs have increased gradually since 2012, the number of RMAT designation requests and granted RMAT designations have grown more rapidly following inception of the program (Figure 4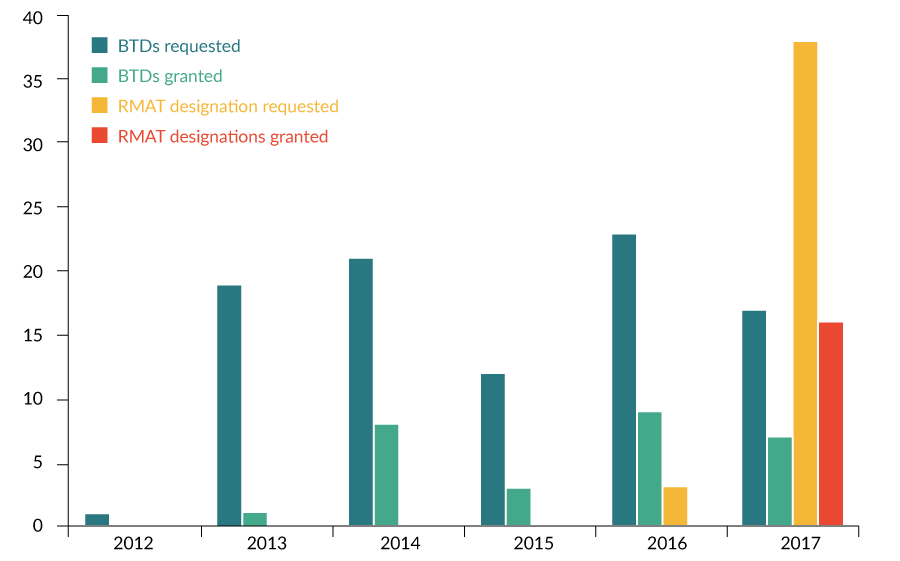
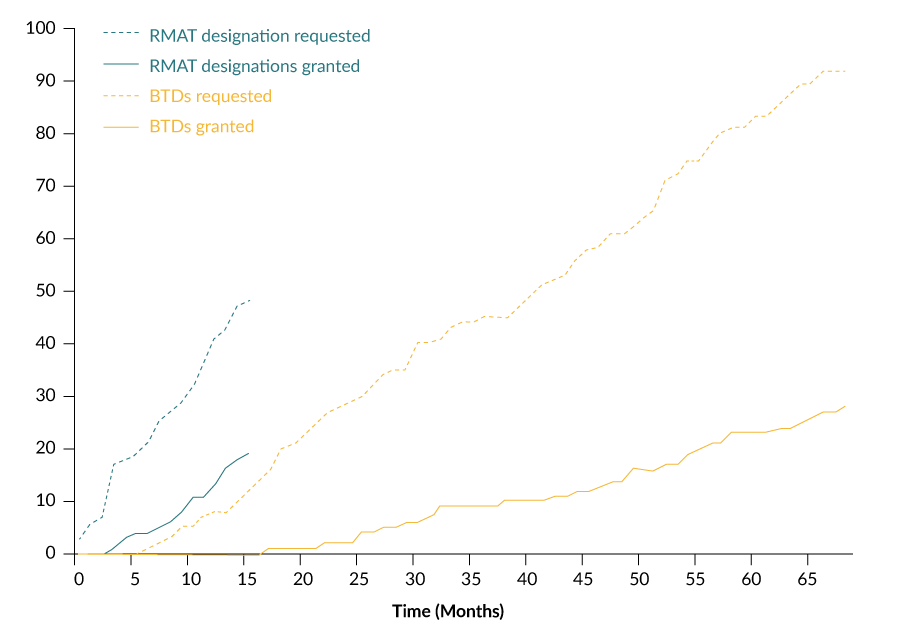
While there was a 16-month interval between the statutory authorization of BTD (July 2012) and the granting of the first BTD (November 2013), there was only a 3-month interval between the statutory authorization of RMAT designation (December 2016) and the granting of the first RMAT designation (March 2017) (Figure 5).
Table 4 summarizes the number of RMAT designation requests and granted RMAT designations by product type. The RMAT program has resulted in designations primarily for cellular therapies, suggesting that it complements the BTD program, which has yielded designations primarily for gene therapies (Table 2). A total of 68% of RMAT designations are for allogeneic or autologous cellular therapies.
Both Table 5 and Figure 6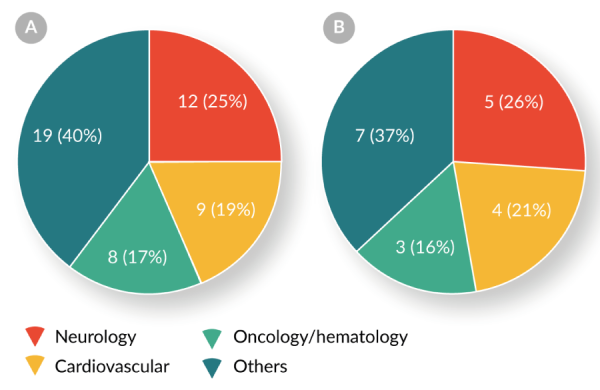
| Table 5 RMAT designation by clinical indication (excluding withdrawn and pending requests). | |
|---|---|
| Products | Granted/requests (%) |
| Neurology | 5/12 (42%) |
| Cardiovascular | 4/9 (44%) |
| Oncology/hematology | 3/8 (38%) |
| Others | 7/19 (37%) |
Two-fifths (21/48, 44%) of RMAT designation requests have been for the treatment of neurologic or cardiovascular diseases. Four products have received both BTD and RMAT designation.These data indicate that the RMAT designation program complements the BTD program by expediting the development of cellular and gene therapies for indications other than oncology/hematology.
The most common reasons for denial of RMAT designation requests have been:
- Administrative issues, such as an inactive IND;
- The original product underwent major modifications during clinical development, such that the preliminary clinical evidence generated using the original product could not be used to support RMAT designation for the modified product; and,
- There was insufficient preliminary clinical evidence to indicate that the product has the potential to address unmet medical needs for such disease or condition (e.g., an early-phase study with inconsistent results among different clinical outcome measures).
SUMMARY
CBER’s Office of Tissues and Advanced Therapies (OTAT) has successfully implemented the BTD program to expedite development of novel cellular and gene therapy products. 2017 witnessed the historic marketing approval of the first such products in the US — tisagenlecleucel, axicabtagene ciloleucel, and voretigene neparvovec-rzyl — all of which received BTD during their development. The launch of the RMAT designation program in 2016 offers a new expedited development program for cellular therapies and certain gene therapies. Our initial experience indicates that RMAT designation will complement, rather than duplicate, BTD in facilitating development of cellular and certain gene therapies for serious or life-threatening conditions. Through these and other programs, OTAT will continue to work closely with sponsors to speed development, review, and marketing approval of innovative, safe, and effective new therapies for patients.
References
1. FDA Draft Guidance for Industry: Expedited Programs for Regenerative Medicine Therapies for Serious Conditions (posted November 2017). Download PDF
2. FDA Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologicals (posted May 2014).
Download PDF
AFFILIATIONS
Xiaofei Wang
Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, Maryland, USA 20993
Wilson W Bryan
Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, Maryland, USA 20993
Lei Xu
Author for correspondence
Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, Maryland, USA 20993
lei.xu2@fda.hhs.gov