
Navigating regulations: novel cell therapy platforms and their path to clinical manufacturing
Cell & Gene Therapy Insights 2021; 7(5), 831–845
10.18609/cgti.2021.083
Manufacturing strategy can have a critical impact on cell therapy development programs, and as regulatory and manufacturing trends evolve, and the need for more efficient manufacturing technologies increases, question of when to introduce novel equipment into a process can pose a challenge for cell therapy manufacturers. Merck KGaA works with a broad range of customers in the area of gene editing and novel modalities and is currently preparing the ekko™ Acoustic Cell Processing System for commercial market entry (Box 1).
In this current pre-commercial phase, discussions with customers have revolved around de-risking, regulatory acceptance, and available documentation. Navigating these aspects as an equipment provider requires very close collaboration with both therapeutic developers and regulatory authorities, and the complexity of introducing such novel equipment into therapeutic manufacturing has sparked many discussions. In the following expert roundtable, industry experts discuss the rapidly maturing regulatory environment, and the critical questions of when to introduce novel equipment, and how to de-risk technologies during therapeutic development.
| Box 1 |
The ekko™ Cell Processing System (Figure 1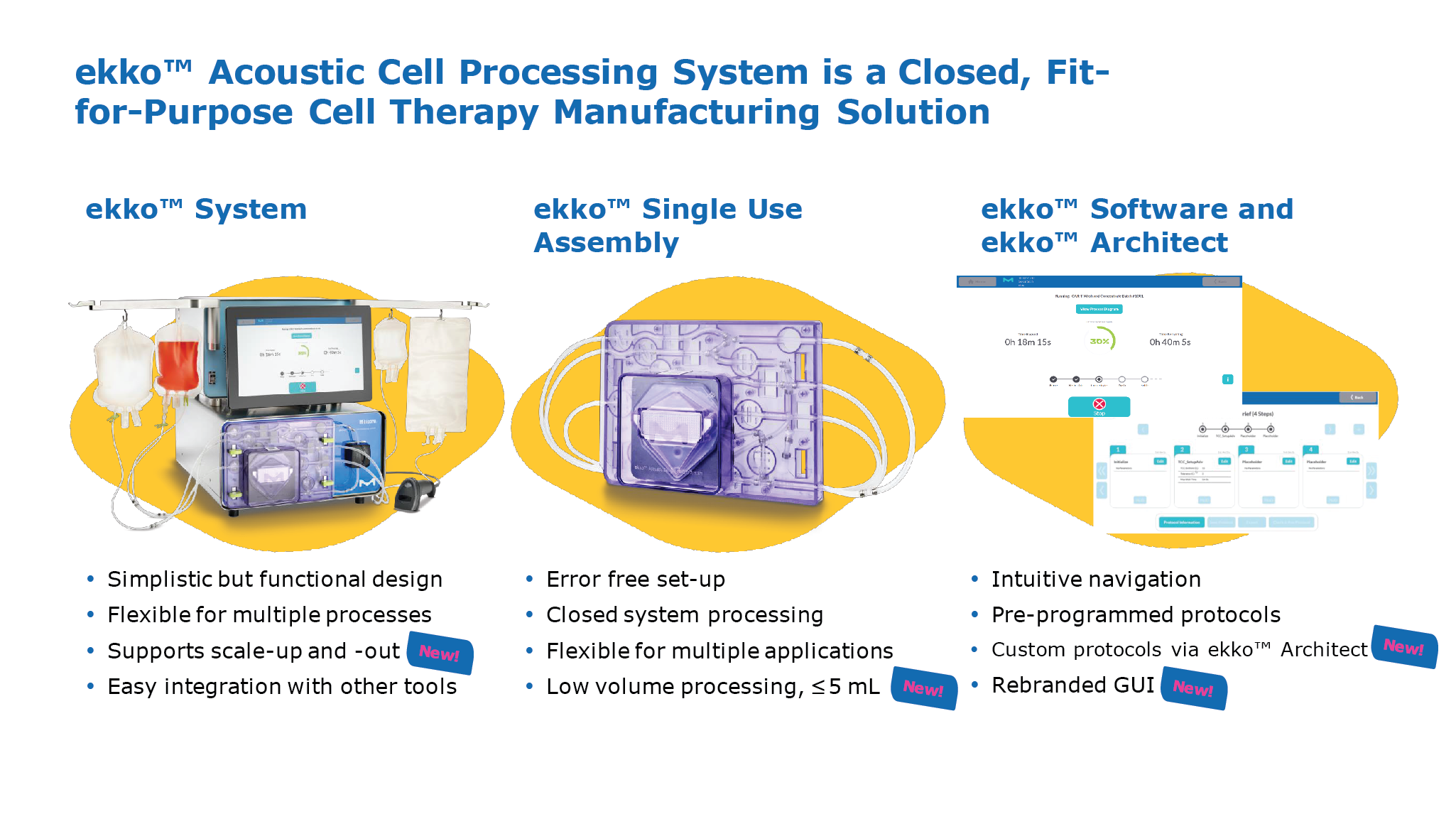 The ekko™ Acoustic Cell Processing System.) is based on the physical properties of a so-called ‘standing wave’. It is generated by a transducer and a reflector, and essentially creates an ‘acoustic mesh’ that can capture and retain cells in suspension, while removing liquids and smaller particles. The approach functions similarly to a traditional membrane filter, but without the disadvantages of a porous, rigid filter material. The ekko™ Acoustic Cell Processing System.) is based on the physical properties of a so-called ‘standing wave’. It is generated by a transducer and a reflector, and essentially creates an ‘acoustic mesh’ that can capture and retain cells in suspension, while removing liquids and smaller particles. The approach functions similarly to a traditional membrane filter, but without the disadvantages of a porous, rigid filter material.The ekko™ System is a closed, fit-for-purpose cell therapy manufacturing solution comprised of benchtop equipment. It comes with an easy-to-use user interface operated via touchscreen, as well as a desktop application, ekko™ Architect, which enables full custom programming. The accompanying ekko™ software supports 21 CFR Part 11 compliance and was designed according to GAMP5 guidelines. The technology has been independently audited, and Merck KGaA is now in the final stages of compiling validation datasets to ensure compliance with GMP guidelines. Given the high flexibility of this technology, it is uniquely suited for unit operations across the entire manufacturing process. Using a CAR T manufacturing process as an exemplar, the system can perform all of the typical wash and concentrate steps, along with media exchanges as required during longer cell expansion phases, as well as buffer exchanges that are required for final formulation. A next-generation system for cell selection, the ekko™ Select, is currently in the final stages of development and will be launched next year. |
EXPERT ROUNDTABLE DISCUSSION




(NB): Welcome, everyone. I have lots of questions for our panel today, and we will also be taking questions from the audience. To begin, we will discuss the regulatory space for cell therapy in general. We have recently heard of more pushback from the FDA, and other regulatory authorities, on investigational new drug (IND) submissions and registrations. There seems to be a tightening of regulations with regards to Chemistry, Manufacturing, and Controls (CMC). Is this something that you would agree with?
AD: We certainly are hearing a lot more about this. At Dark Horse, we hear from a lot of people who have that hypothesis, if you will, that things are tightening up, and that cell and gene therapy is receiving an increased level of scrutiny.
We actually disagree with this hypothesis. We believe that the overwhelming majority of what is happening in the regulatory dialogue is that the field as a whole is shifting towards the commercial end of the spectrum, and away from the early-stage developmental end. This part of the spectrum has always been tightly regulated, with relatively little flexibility around the GMP guidelines that the FDA and other regulatory bodies provide.
For example, since we all last spoke while preparing for this discussion, Iovance has further delayed their BLA submission. This is the second successive delay and will make their cumulative delay at least 18 months. Almost all of this delay seems to revolve around their potency assay.
Last time I checked, a fully validated potency assay has been an absolute requirement for any biologic since the inception of the Center for Biologics Evaluation and Research (CBER). There were some very choice remarks from Peter Marks on this subject the other day, which I interpreted as an expression of irritation and frustration with the field more than anything else. I have the quote here:
“Pick something, pick some quality of the cell, pick something you think might correlate, and measure that. We’ll take any offers that are reasonable.”
Our position is that the FDA is losing patience because the field is entering a space that has always been strictly regulated. We see this as an increasing threat to the field.
(NB): Natika, you deal with both the viral vector space and the cell therapy space through the equipment lens. Any thoughts on what Anthony just described?
NC: I think there is an increase in expectations, given guidance that has been put out for manufacturers. I do agree that these regulations have been written and they are there for us to follow. The expectation is that we catch up and provide all of the data that is expected.
Speaking from being inspected as a viral vector manufacturer, the expectation on risk assessment is evolving. We have documentation and risk assessment for the most key and critical pieces you would expect – for example, how you mitigate cross-contamination – and that has always been there. But there are so many other layers now around risk assessing numerous parts of the process, numerous equipment additions, and having documentation around all of that, and also the need to have it put in a formal package. For example, the requirement to have a contamination control strategy where all of these things are put together in one place. That is very handy, but that language wasn’t written years ago. We have been in the same location since 2004, and these things weren’t written.
When I think about how regulators will put out guidance, it is generally because they are seeing that the industry may need some help so that we are collectively submitting the same things, and there aren’t gaps from one sponsor to the next. The difference I am seeing is that expectations on risk assessments are much broader, and we need to be much more detailed.
(NB): Matt, you are on the therapeutic developer side of things, and deal with a lot of supply chain questions. Any thoughts from that angle?
MM: From a manufacturer perspective, and considering path to market, I think there is a really traditional trade-off here of speed versus a more robust manufacturing operation, and trying to build the best product possible.
The promise of allogeneic cell therapy is that it offers a reduced timeline to make a difference for patients, and the opportunity to potentially commercialize products after approval from a pivotal Phase 2 with positive clinical data.
As a result, process improvements or enhancements, such as closing a process from an aseptic manipulation standpoint, or dual-sourcing key materials, can be compromised on or may have to represent that trade-off, and then the timeline is condensed. But this is not a new trade-off.
It does need a risk-based approach, as Natika mentioned. What is unique to the cell therapy space from a regulatory perspective, and maybe furthering Natika’s point, is that you have to apply those same principles across a much broader spectrum, and in particular this applies to starting materials. This is where the evolution or maturation of regulatory principles is still coming into focus as it applies to vectors, mRNA, plasmids, donor cells, and so on.
The old adage that everyone wants to be first and fast, but nobody wants to learn the hard way, is probably more applicable than ever in the cell therapy space.
NB: To follow up on that, you are also in charge of evaluating equipment. In cell and gene therapy we have been repurposing existing equipment from the blood industry, and often relying on technologies that are mainly used in the research space. This is one of the areas where we find ourselves in a bit of a gray zone, where we as an equipment provider are trying to develop material and equipment that is GMP compliant, and specifically designed for the cell therapy manufacturing environment. This is a bit of a shift for a lot of our customers when bringing in completely new technologies like acoustics.
(NB): Matt, from a therapeutic development side, and with everyone pushing as hard as they can to get to market and treat patients, any perspectives on how you make decisions on when to introduce, and how?
MM: First and foremost it starts with the technical capabilities of any piece of equipment. At Allogene we are fortunate to work with a very talented research and process development organization that can assess those capabilities.
Leading the sourcing and supplier management function at Allogene, I have a vested interest in ensuring that supply chain has a seat at that table, and is thoroughly involved in the process to look for particular pitfalls.
In the cell therapy space there are two key areas regarding commercial setup and expectation of some of these suppliers, as well as the management of their IP and confidential information. Here I would point to a traditional supplier-manufacturer relationship that most of us are used to in this space, and to the example of bioreactors and monoclonal antibodies (mAbs).
There has always been an exchange of confidential information for the sake of process optimization, and in some cases, comparable information around how the equipment works is not readily shared with manufacturers today in the cell therapy space. The space is certainly new and developing, and as a result, there is novel IP, but manufacturers don’t have any interest in this confidential information beyond the purpose of improving our process to make the best products for patients. This is no different than the mAbs and bioreactors of years prior.
Unfortunately, this setup means that process development and characterization can be hindered, but it is my strong opinion that this will not last in the short- to mid-term as competition increases. Traditional relationships will return, and suppliers will be sought that help manufacturers create the best product – period.
For now though, when considering new partners in this space, it is paramount to ensure terms and conditions around commercial expectations are clear and reasonable.
(NB): It is definitely my opinion that providing the broadest access possible will ultimately help everyone. Natika, as I mentioned earlier, you have two hats. You have responsibility for our gene therapy and viral vector manufacturing business, where you have brought the facility to commercial approval. Any thoughts or experiences from that perspective?
NC: We have had the benefit of going through the process of becoming a commercial manufacturer, validating and implementing all of the equipment in use, and sharing that and explaining the depths of those qualifications to the regulators. We have been successful in that.
One of the things that now is a benefit to us a few years later is that for all of our clients, who are for the most part in their clinical phases, we are prepared, as their manufacturer, for that commercial endpoint that they are looking towards. We already have these things in place in terms of our facilities and our process control expectations, and we bring those to benefit the client.
However, the expectations for early-stage clinical manufacturing aren’t the same, in terms of validation. We deliver interim levels of support based on phasing, but we are prepared, and we provide that roadmap to commercial success.
In terms of a case of novel equipment that we have implemented, we have had equipment where we had some failures in the earlier stage and had to go back and work with the supplier of that equipment to map out and make sure we had taken care of failure modes. As a service provider, we would do that on our own proprietary equipment, but we have also brought in equipment that clients requested to use in their process, and then we helped them navigate through making sure that could be qualified sufficiently for consistent processes.
(NB): We see that every day in the CDMO space, where we have very bespoke equipment and that needs to be managed. Anthony, to speak to your point earlier on everyone gunning for the endgame of commercial, and also picking up on what Natika just said about regulations not being as tight in the early stage: people often use the term phase-appropriateness. If you think about working with your clients, what recommendations do you have on how to go about that?
AD: It is a very important phrase to bear in mind, because obviously you are filing an IND or a Clinical Trial Authorization when you are at that point, not a Biologics License Application or a Marketing Authorization Application.
There is phase-appropriate, and there is phase-appropriate. There are certain things you do in early-phase that you will live with forever.
I will go back to the potency assay as an example of this, then I am going to circle through to equipment. The potency assay does not need to be validated for your IND, your Phase 1, your Phase 2, or even – if you are foolish enough – your Phase 3. But there is an expectation that it will be validated when you go for the ticket.
It is phase-inappropriate to develop a validated potency assay for your IND. However, if you get to the endgame and you discover to your surprise that your assay is not validatable, it turns out not to relate sufficiently to your mechanism of action, or it turns out not to be the critical quality attribute mirroring the CQA you thought it would, then that is a problem. You either have to have an abundant supply of retains to test everything that has gone in the clinic previously with your new potency assay if you are going to have to replace the old one, or you are going back into the clinic. That is a very tough thing to say, but that is how it is. There is no time machine to go back and repeat and re-release lots if you have to switch an assay.
That is a good example of what is and is not phase-appropriate. The same thing rattles through to equipment: if you have manufactured your early-stage material with a piece of equipment that then turns out not to be commercial-grade, then the early-stage material’s quality will be called into question and there will be a vigorous discussion of that with any regulator.
I have to say, it is so refreshing to see brand new technology like the ekko™ System coming into the field. It is quite rare, and I think more events like this will occur, and need to occur, to bring down the cost basis of these products and increase their quality. But the biggest pitfall is doing your earlier stage manufacturing one way and then landing yourself with a nasty big comparability issue further down the road.
(NB): The question of supplier qualification and validation feeds into providing our customers such as Matt and his team, with the right documentation. Natika, with that second hat you wear where you support us in qualifications, validation, and documentation, how do we go about this? How do we support and ensure customers get what they need?
NC: We have development teams that are working on not just equipment, but platforms, and we are looking at how to improve our processes every day.
For example, suspension cells in vector manufacturing: most of the processes now are in adherent cells, and so the early approvals are based on adherent cell processing. That speaks to Anthony’s point that when you have developed all this data, you don’t want to have to go back with a huge comparability issue at the end. For a client to start with an adherent cell process and then get to market with a suspension cell process, that is going to create issues and a big delay.
We are not seeing that the first in the pipeline gets the change that has the new equipment, for example. It is not the folks that are far ahead in their trials and have locked down their process, it is those with early stage pipeline candidates, or more broadly those earlier stage companies, that we can talk to and say that we have a piece of equipment that is going to really speed up the process, and we also are supplying validation that goes along with that. I’d point the audience to our Emprove™ Program, for example.
For a lot of these novel therapies we are using research-use types of equipment. We will then proceed to having a conversation with regulators that this is the only equipment we have that serves this purpose, and it is not available from the supplier as a fully validated, mature piece of equipment. For our own equipment, such as the ekko™ System, which is not yet fully validated, we help by putting together the plan for that piece of equipment. Our approach is to work with the customer and provide as much data on this equipment as we can, from whatever research use that we have, and provide expected timelines for when we will have datasets available for a fully validated system.
So, there is a spectrum where we tell customers that we know that this is the equipment they need to use right now, and in the future, for their next candidates, we have suggestions for them. We are working early with the customers who are developing the technology, we are committed to working with them, and we are working together to get to a GMP compliant piece of equipment. There is an expectation of what it needs to look like commercially, but we have been able to work with the agencies and say it is not quite there yet, and they have been accepting of that. As we are now maturing, we are really trying to bring in equipment that is as far along on that path with all of the qualifications and validations that we can get, and offer that.
(NB): Anthony, any thoughts on how you have supported clients in bridging the gap between us as suppliers, the therapeutic developers, and the regulatory authorities?
AD: The best and only way to do it is with really open channels of communication.
There are bad equipment manufacturers, who will remain nameless, who blackbox stuff. There is a bit of paranoia about trade secrets. But this is a field where, in a very real sense, a rising tide lifts all ships. Likewise, the regulators do not like surprises.
There is a lot of game theory we see from very seasoned regulators, and we see it from law firms too. We get involved with compliance and remediation cases where people have already lawyered up. The lawyers are good, and half of them are ex-FDA themselves. But there is a tendency for game theory, and an attitude of “we don’t want to tell them this because they might not like it, and it might sow problems down the road”
It is one thing if you are in a pre-approval inspection from the FDA Division of Manufacturing and Product Quality (DMPQ), because those inspectors are in uniform, and have no sense of humor and lots of clipboards. But if you are still dealing with the scientists and the medics at the FDA, I think they enjoy proactive and honest upfront communication. They thrive on it. They see things that you do not know they have seen, and they cannot supply you with those pieces of information, but they can use them in their response to you.
Bring more information rather than less to regulators – and here I am talking to the therapeutic developers as well as to your people, Nina – and you will be surprised how helpful that is in the long run.
NB: To add to that, one of my experiences in a pre-Merck KGaA life, when I was promoting and working on a new piece of equipment, was exactly that. I once gave a presentation with FDA people in the audience; they approached me afterward and were extremely excited about the equipment. They invited me to come in and talk about it, and bring them up to speed on all the new stuff that is coming up. From a supplier perspective, that was a really interesting experience for me and something that I have always valued very much.
(NB): Matt, you are on the end-user side of things, and you have seen things come through that are research-use-only, but serve the purpose to a tee. How do you support your internal programs when dealing with the likes of Merck KGaA, or other equipment suppliers?
MM: We made the earlier reference to phase-appropriateness, and this certainly can occur if the roadmap exists. Suppliers that have the right experience can be extremely valuable in that capacity.
Within Allogene we have a common saying: “right size for right now.” As a company, we need to continually check ourselves to make sure we are staying at the forefront, but also that we are not out over our skis, as we don’t have unlimited resources.
In that regard, phase-appropriateness in this space, if you have the opportunity to potentially commercialize in Phase 2, is a bit of a myth. It is really best to start with suppliers who have been there before, even if it is in other modalities. They know what it takes for a product to be commercialized, and this is where a risk-based approach comes in again and is so important.
Working with some of the niche suppliers in this space that have really exciting technology, they certainly don’t intend to have lower quality or regulatory standards, but it is absolutely a risk. Larger organizations like Merck KGaA, or some of your competitors, can step in and show their value and experience here, particularly as it applies to equipment or reagents. On the equipment side, I would call out validation packages, software, and hardware in particular.
(NB): A question has come in from the audience that is perfect for this line of discussion. When making automation and process closure improvements post-approval, is it generally advisable to group as many of these process improvements together as possible, to make comparability studies and associated regulatory dialogue more efficient?
AD: So basically should you batch up your CMC amendments, or drip-feed?
There are pros and cons, obviously. The drip feed sounds worse than it is, and enables you to bring amendments in a timelier manner to the agency. The one-at-a-time approach is riskier because if you put in six amendments, statistically there is a bigger chance that something is going to trip up. When you have got all six bundled together, and if number five of six is the problem child it is going to hold back the rest of the class, potentially. On the other hand, if you batch them up, it is a more complete up-rev, and if there is comparability to be done, it is only one set of comparability.
Another argument for the drip feed approach is that time is money, and if you have got an improvement to the process, getting it into the plant that is spitting out either your commercial drug or your pre-commercial drug faster is a good thing. On the other side of the coin, batching does reduce the regulatory burden, and it does make things more efficient.
For truly significant amendments, which require deep comparability, I think it is important to get them in front of an agency as soon as possible. For lots of little ones, or LOLO as we call it here, batch them.
(NB): A very broad question that essentially goes back to the overall regulatory environment – do you think that international harmonization of requirements between various regulatory agencies is a strong trend, and would such harmonization be more viable in specific disease areas? Anthony, any thoughts on international harmonization?
AD: We recently hired Don Fink from the FDA, and in the last 5 or 10 years he has been involved in a lot of harmonization discussions between the FDA and other regulators. We also have a lot of clients for whom we often take the US material to the MHRA, and we take the EMA trials to the PMDA in Japan.
It is a strong trend, and it is increasing. There are great things around the world, like the EudraLex and the International Organization for Standardization. It is increasingly international, and I think this is the way it is going to go.
Especially with the COVID vaccine situation. It is kind of nuts to have the MHRA approve a vaccine first, out there in London, then the FDA with a very fast second, and the EMA just struggling, frankly. There is no need for that, and I think things like the pandemic will just reinforce that trend. These products are either gene therapies like the AstraZeneca and J&J vaccines, or basically a gene therapy, like Pfizer and Moderna. I think that will reinforce the trend. I would say it is a strong yes to the question.
NC: I echo the strong yes.
The good news is that as our clients are submitting their BLAs, or getting new products that they want to get approved; the agencies are asking us first for our inspection reports. They want to see which agencies have inspected the site, referring to our viral vector manufacturing facility. The first question, for example if we have had the FDA come, is can we see your inspection report? Other agencies are definitely looking at those and making acceptances without necessarily coming to do an on-site inspection themselves.
I will also bring up COVID. In 2020 we had several inspections from different agencies planned. Of course, restrictions for travel, for coming on-site, for the safety of our operations, and having a 2-week quarantine for regulators to come from another country and sit in a hotel for weeks before coming on-site, just made everything entirely unfeasible.
They looked at what the other agencies had reported on before, and we were able to move forward, or have extensions, and so on. That was inter-agency, and is a direct benefit of the harmonization that is happening.
MM: Maybe it goes without saying, but obviously manufacturers clearly benefit from that harmonization. As much as possible, trying to follow a single set of clear guidance is an extreme advantage, especially as some of the players coming up in this space are smaller companies, and quite frankly don’t have the resources to look at regulatory guidance from many different places.
As much as that industry trend continues, it significantly helps the industry as a whole to push these therapies from the clinic to a commercial setting.
(NB): Another audience question – Daniel from Allogene is postulating that some equipment manufacturers have been less than transparent regarding operating ranges that are embedded deeply in recipes, and this is creating a validation and process troubleshooting challenge. Do we as the panel see this changing in the future? I can only speak for us, but we try to be as transparent as we can. We are creating our documents in such a way that there is as much information as we can provide, and create the datasets that we share with all our customers that we work with. Any comments from the panel?
AD: Daniel, we must have been dealing with the same equipment manufacturers!
It is exactly what Daniel has articulated here, a classic example of the sort of thing I was mentioning before, where people are a little bit Wizard of Oz about their equipment, and want to keep it blackbox.
Everyone is going to try and bring out competing products. There will be competition to the ekko™ System, either literal competition that will end up in IP court or similar, or non-literal competition where some other orthogonal physical method will try and eat your lunch. Fundamentally, competition is a good thing, and cream rises to the top. It doesn’t help in the long run to take a short-sighted view.
(NB): Another question that tags on to that one by talking about the adoption of new equipment, automating processes, and speeding things up. This has most likely, or pretty certainly, happened in the mAb space in the past, which has got us to where we are right now where monoclonal antibodies can be manufactured in large quantities and at a very reasonable price. Learning from the mAb space, how does the panel think this might play out for cell therapy? Will adoption speed up and innovation slow down, or will there always be a mismatch between the two?
MM: This is a really challenging question, and maybe from a scale perspective we will always have that challenge.
We talk frequently in the industry about scaling up versus scaling out. Many of the processes in CAR T manufacturing today can still be very much be characterized as benchtop manufacturing scale. It is essentially a research-scale operation that we are scaling into a commercial phase.
Considering what we do in mAbs today, with 2,000 liter bioreactors, or even much larger, I don’t see that as being a driving trend in the near term for cell therapy. I see it more as a scale-out process that we would follow. From the standpoint of a manufacturer working with suppliers, the point we discussed before regarding an openness on how we make the process the most efficient, and deliver the best product for patients, needs to be the primary goal and focus.
NC: I have one thought regarding this mismatch of having the right technology, or the most efficient technology, and the speed that we need. A direct example that we have, because we also have that process development side, is that we have looked at new client’s processes, for example, and recommended certain changes, and done studies for them to translate their process into our facility with X equipment.
What we have seen in the last three years or so is that the clients have been, and still are, a little bit hesitant to change things when they are on their path with their first candidate. Part of that mismatch is that they have told their investors here is what our process is, and here is what we are doing when. We come along and make suggestions and they say yeah that is great that we could get better yield, but we are sticking with what we have got. I think there is still going to be some of that.
(NB): The last question today goes back to scale out. How do you see the role of a modular cleanroom facility with a quality management system (QMS) in relation to its operations, in helping to expedite its biotech client’s cell therapy development pipeline? The facility is not a classical CDMO, but one that provides the necessary manufacturing facilities for multiple small and medium enterprise (SME) companies at the same time.
AD: We like them. There have been pioneers in the field, and I think there are now a lot of fast followers.
It is a spectrum. There is the idea of the manufacturing facility on a truck and the idea is not as dumb as it sounds – the blood donation industry has been just fine doing this for decades.
You can have modular: “QMS in a box” if you like. You can have semi-modular, where the walls and ceilings and floors arrive on a truck and they are assembled IKEA-style inside the aircraft hangar of a building, and there are a lot of facilities around the world that have basically done that. They partially come with an associated QMS; they certainly come with their own facilities, utilities, and metrics.
There will always be good old-fashioned stick-built and purpose-built, and so on and so forth.
Looking at the question carefully, the part I disagree with is saying that that facility is not a classical CDMO. It doesn’t really matter that much. As I have said on previous forums, we sometimes see the facility as being more the lumen of the two. That is what is under GMP – in a truly closed system, the facility is inside that closed system. The box, the room, the pod, whatever it sits in, is somewhat on the periphery of where regulatory focus is going to be down the road.
NC: In my experience, over some decades of coming up with ways to manufacture these cell therapy products without the huge expense, having something that is modular has been a very useful answer. They have helped bring things to where they are today.
Sometimes we are looking at aseptic processing, and at whether to have a closed system. Those are considerations we have to have, and it is easier if you have a closed system. They are helpful, because if you can get your batches made, and you have some data you are collecting, when you get further along and you move to other pipeline candidates, this is data that is necessary. Talking about modular cleanroom facilities, those do meet the requirements.
Obviously, I like our purpose-built facility and our years and years of experience, but it is certainly a feasible way to go.
(NB): Matt, as a therapeutic developer, any thoughts on whether you would go with that for your manufacturing?
MM: There are really good arguments for both modular, cleanroom-in-a-box type solutions, and more traditional stick-build. I think is a worthwhile debate today.
Speaking from Allogene’s perspective, we have just completed our own internal manufacturing facility. The need to have control over the operation at this level and stage in development is paramount. It is less about the design of the facility, and more about ownership of that and being able to oversee the key operations we see as an important lever to success.
NB: Thank you to everyone who provided questions, and a very warm thank you to the panel.
![]()
Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: AD declares that Dark Horse Consulting is a consulting group that serves the Cell & Gene Therapy Space whose clients are confidential. NB is an employee of MilliporeSigma and a member of the Scientific Advisory Board of SMART, Singapore-MIT Alliance for Research & Technology.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2021 MilliporeSigma. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is a transcript of a previously published webinar, which can be found here.
Webinar published: May 27 2021; Publication date: Aug 11 2021.