Fitting product to process: raw materials customization for cell therapy manufacturing success
Cell & Gene Therapy Insights 2021; 7(2), 183–198
10.18609/cgti.2021.029
Cell therapy manufacturing involves highly complex processes, with large numbers of inputs, and therefore a high amount of associated risk. A robust supply of high quality raw and ancillary materials is crucial to successful manufacture of cell therapy products – but selecting the best supplier, and the right product, can pose a challenge. Raw material customization may require more up-front investment from manufacturers, but even relatively small modifications to packaging and fill sizing of off-the-shelf materials can provide cost-effective products that better fit process requirements, and help to de-risk manufacturing. Identifying risks up-front and customizing products where required can save time, money, and ultimately speed up commercialization of therapies.
Small changes that can translate to big benefits
Case study: A simple fill modification of an off-the-shelf reagent
When considering customized raw materials, even small changes can help to drive a more streamlined manufacturing process. Simple fill modifications of off-the-shelf products can help to manage risk, improve cost, and save time in the clean room.
In this case study, a manufacturer wishes to add an optimal quantity of cytokine to media used for culturing cells. Introducing this reagent into the process is often an open step. Cas cytokines are commonly supplied lyophilized in glass vials, while the media required for closed or automated cell culture is provided in bags. The use of predefined ‘off the shelf’ sizing also means the quantity being added may need to be modified manually for each manufacturing run. As cytokines are a reagent of biological origin, further complexity is added, as there may be lot-to-lot variability. This can result in the need for lot-standardization efforts in-house.
As illustrated in Figure 1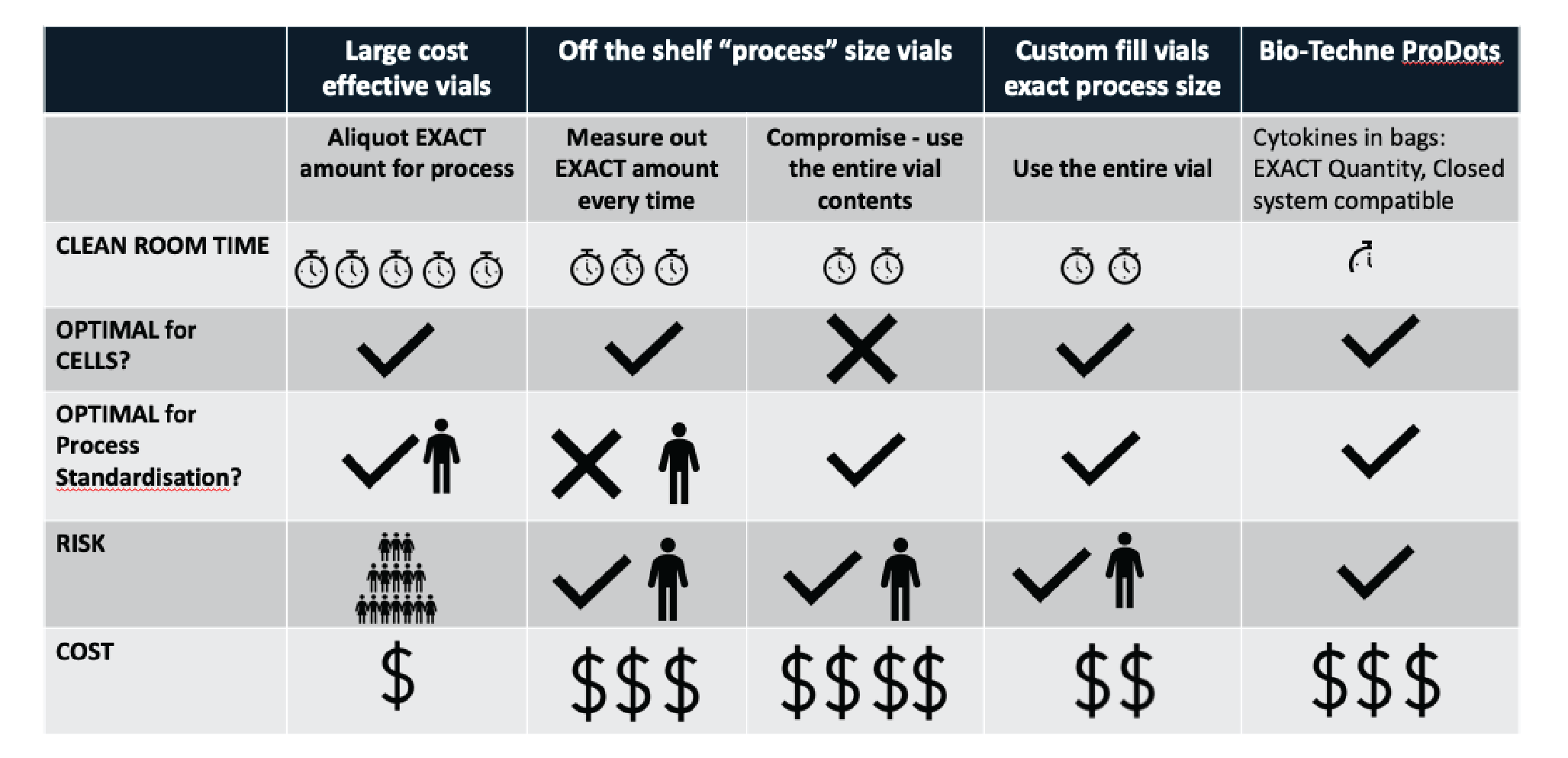
Option #1: Take a bulk quantity of cytokine & aliquot the exact amount for the process
This provides the optimal predetermined quantity for the cells, and is optimal for process standardization as the same operator is measuring each aliquot. However, this also introduces risk – if the cytokine is contaminated, or resuspended in the wrong volume, multiple patient products could be affected before the error is discovered.
Option #2: Off-the-shelf ‘process size’ vials
This option carries less risk as the vials are all coming straight from a supplier, and will be sterility tested. With no pre-aliquoting required, this approach will also save time. However, it may not be optimal for standardization between every manufacturing run. It is also possible to use a process size vial to oversaturate the culture, to eliminate potential measuring errors and improve standardization. However, this may not be optimal for the cell culture.
Option #3: Customization of fill & packaging
If suppliers provide the cytokine aliquoted at the exact requirement of the manufacturing process then it is both optimal for cell culture, and more standardized. If that packaging could be further streamlined to being closed system compliant (e.g. Bio-Techne ProDots [1]) then the risk of having an open step is removed. If manufacturing is being performed on a significant scale then bulk savings, alongside reduced risk and cleanroom time, will balance additional costs.
Bespoke products for specific needs
Modified off-the-shelf products may be suitable for some manufacturers – but in some cases, the product a manufacturer needs does not exist. The following case studies highlight the important factors to be considered before committing to a critical reagent that may not be appropriate for future clinical development.
Case 1: Licensing & freedom to operate
The project
A client has been using a conjugated and hybridoma-derived Research Use Only (RUO) antibody for all of their preclinical work. Now, after risk assessing they have determined they are unable to move this material into Phase 1.
The problem
The product is not available in GMP grade, is not manufactured under animal-free conditions, and use is restricted by licensing issues.
The foremost consideration is whether the client has Freedom to Operate using this clone, as the owner of the clone may have sublicensed it to a different vendor, or there may be restrictions on commercialization or modification of the product. Having an open dialogue with the vendor to ensure the material is suitable for the intended use, prior to committing to a specific product, is imperative. In addition, the future conversion of the product to a recombinant clone, or being able to manufacture it in a GMP facility that is also animal-free compliant, will be important factors from a regulatory perspective. If the product comes into contact with animal-containing components, this could put approval of the therapy at risk.
In this case the vendor was not only unable to manufacture under GMP conditions due to their capabilities, but their license also restricted their ability to do so. These issues may not be addressed during the usual purchasing process – but giving a vendor the ability to understand where a project is going, and providing them with information on a client’s ultimate goals, will allow client and vendor to work collaboratively to ensure scalability and success.
The solution
A licensing conversation with the owner of the antibody clone led to them agreeing to a contract that allowed the material to be modified, converted to GMP grade, and supplied to the client at the scale required.
Case 2: Scaling up for future manufacturing
The project
A client is using a contract manufacturing organization (CMO) for RUO manufacturing of a proprietary protein sequence used in therapeutic discovery.
The problem
The current manufacturer is unable to scale the process to meet required yields, and cannot develop under GMP conditions.
In this instance, licensing is not an issue as the product is a property protein sequence being used in therapeutic discovery by a clinical development company. The company had outsourced manufacturing to a CMO that was able to provide a pure and active protein product which worked in all of their preclinical processes. However, once the company was ready to move into Phase 1, they approached the vendor to convert the protein sequence to a GMP product, and found the manufacturer was not able to meet their requirements.
These issues are frequently caused by the need for animal-free conversion of the manufacturing inputs while also scaling up to the multi-gram yields that are required, and the need to manufacture under GMP conditions.
From the client’s perspective, asking whether or not it is possible to convert a given RUO product to GMP, or even being involved in the RUO development process, can help with the transition from preclinical to clinical work. However, many vendors will not allow input and transparency around their internal processes, or allow modifications to their processes. Finding a vendor that is able to meet manufacturing needs, and be flexible in how it fulfils those needs, will help with commercialization, future forecasting needs, and future supply runs.
In addition, regulatory and quality support are key when a company reaches the stage of submitting an IND and filing other required documents with regulatory bodies. If there are differences in how they test with validated assays, or if they are unwilling to share details about the manufacturing process, a client can find themselves in a situation where they have a product that works, but the documentation regulatory bodies require is not available.
The solution
The product was taken in-house (at Bio-Techne), and all processes were converted to animal-free and GMP grade. Appropriate regulatory documents were filed to support the client throughout development.
Case 3: Continuity through clinical development
The project
A client is using a RUO protein and RUO antibody from different suppliers during preclinical development.
The problem
Supply chain and manufacturing inconsistencies between critical reagents.
This case study highlights the importance of supply chain continuity to support and simplify clinical development. At the point that a product has been developed, and multiple products may be going in to a manufacturer’s workflow, difficulty can arise in managing several different vendors. This particular client had been purchasing RUO antibody and protein from two separate suppliers during their preclinical development, and chose to condense down to one vendor that could supply both in a GMP fashion.
The main problem the client faced was supply chain and manufacturing inconsistences between critical reagents. The need to coordinate deliveries, and coordinate and align the quality systems that were used during manufacture of the different materials, was leading to concern about the quality of the final product.
Aligning the quality requirements between different vendors, and comparing them at an early stage, may be challenging. In contrast, selecting one vendor and ensuring they provide all the quality requirements a manufacturer needs for one product can make it easier to then meet requirements on additional products.
In addition, when developing a custom product, the stability of the raw materials used can potentially affect the final process. Off-the-shelf products typically have stability studies performed on them so that the manufacturer is aware of their shelf life. With a custom product, it may be possible to instead establish extended stability that is aligned with the shelf life of the intended final product. In addition, the client can specify the assays that are performed to better optimize the stability studies.
The solution
By condensing suppliers, it was possible to modify the certificates of analysis, and modify the post-vialing QC testing, cell line testing, and other analytical testing, either in-house or outsourced. This simplified both the client’s workflow, and the documentation processes when they came to file for an IND with the FDA.
SUMMARY
Investing in the selection of raw and ancillary materials early on in the manufacturing process can save significant time in the later stages of development – not only does it allow for improved forward planning, it can also enable the development of tailor-made product that better meet a manufacturer’s process requirements.
Customization options can offer more flexibility from off-the-shelf products that already exist by providing specialized bags or filling sizes, or specialized testing. Alternatively, developing a de novo product can provide a bespoke solution for the often complex requirements of cell therapy manufacturers.
Q & A
Q What are the main issues that each of you currently face in sourcing and securing an adequate and continual supply of your critical raw and ancillary materials?
RL: One of the most important things is the pace of innovation in cell and gene therapy. The industry is maturing very rapidly, and has gone from an academic dream just over a decade ago, to commercialization today. It is starting to outpace the ability of off-the-shelf materials to accommodate it.
There are two different reasons for that: the science, and the compliance. From a science perspective, what we are looking to do as we innovate is to bring something from the benchtop to the bedside as quickly as possible. Oftentimes in these kinds of atmospheres, we are finding something potentially from academic papers, from research, and we are trying to bring that into patients as quickly as possible. We also want to do it in a safe and complaint manner. This means we need to find a partner that can help bring these innovative materials from RUO to GMP. We will often have shifting specifications, or shifting needs, as we discover more about what we are going to do with this material.
On the compliance end, again as we mature as an industry and are approaching commercialization, it behooves us to increase our compliance and increase our efficiencies, by closing the process and making things more efficient in general. One of the issues with off-the-shelf materials is that often a lot of the materials that the industry had used previously were essentially holdovers from blood bank processing, and things like that. They don’t fit the process very well. We are trying to find materials that fit the process, and often that will require customization. We have to find the right partner to do that with.
LB: In my experience, with the cell and gene therapy industry being in relatively early stages, often a lot of these materials are only available from one supplier. These single source materials present the greatest potential risk to your supply chain.
It is therefore incredibly important to develop a relationship with your supplier as early in the development process as possible. As the user, you need to communicate to your supplier what your needs are and provide as thorough an overview forecast as possible. This way, the supplier can either confirm that they can or cannot fulfill that need, or alternatively they can begin to develop their own internal manufacturing capabilities in order to meet your needs.
JT: One of the key considerations at the top of my mind, especially with the pandemic and how important business continuity is, is when you think about ensuring you have the appropriate level of safety stock, make sure you work closely with all your different suppliers. Even when you think about the significant demand last year with masks and gloves and things like that – it is about really looking at your overall business continuity strategy. That is a critical component we need to think about to ensure we have the critical raw and ancillary materials needed to meet the demands of the industry.
Q Looking at how to then mitigate those risks and challenges, what are the key tools at your disposal for managing risk in your raw material supply chain?
LB: You always start with qualifying your supplier and performing routine audits, ensuring that they are manufacturing at the appropriate GMP level, and that they have cross-contamination controls in place. You can ensure that their raw material specifications include some identity and safety testing.
You also want to try and identify an alternative supplier – you should at least try to have a dual source, at a minimum, for each of your materials. Creating a custom material with an alternative supplier is a great way to avoid risk to your supply chain.
You can use a risk assessment that includes supply chain risks to identify those risks, and implement mitigations at the supplier, or through your own internal testing. Then you will have your internal verification testing prior to manufacturing, and at a very minimum you need to include identity and safety testing.
As you move through the development process, you can add on more critical attributes as you become aware of them and as your process knowledge and material knowledge increases. In addition, keeping up to date with what others in the industry are doing, and what regulatory documents and guidances are available, will certainly help you reduce risk in keeping your supply chain.
Finally, it is all about your procurement. Having a really good relationship, having routine meetings with suppliers, and conveying any changes in your supply chain or demand are key. For example, can you give your forecast 9 months in advance, versus three? And of course if it is a larger manufacturer, ensure you have multiple manufacturing sites qualified and not just one location, for example.
JT: I would add that I really like the idea of secondary sourcing. That is a key component to mitigating risks to your overall supply chain. Something critical to that as you think about the risk to your supply chain is quantifying with each of the different suppliers the possibility of failure.
When you think of the overall materials that are needed, there are multiple different suppliers that companies are working with to produce cell and gene therapy products. It is about understanding where the highest risk is, and deciding what to focus on first. That will be a key component of building out an overall strategy as you think about supply chain risk mitigation.
RL: Something I think is really important, and a great tool, is the partnership between the supplier and the company. You want to be very transparent about what you need, how much you need, and when you are going to need it – especially in autologous cell therapies, where we are predicting the number of patients we might get that year. Let the supplier know there is some level of uncertainty in those patient level forecasts, and build those in to the supply agreement so there is transparency on both sides. This means that each side knows what kind of demand they are going to expect, and allows you to ensure that the supplier can commit to that. Transparency on the ability of both sides to supply for these autologous patients is crucial – each lot is a single patient, so we have to make sure we are able to do that.
Q Mitch, we have heard a great deal here about the importance of partnership as being central to managing risk – from the perspective of the supplier, would you agree with that sentiment?
MB: Whenever a supplier can plan ahead, and potentially sequester lots specific for that therapy, or manufacture additional material according to what is forecasted, that is always going to help streamline the process. As Raymond said, it is an estimation of how many patients you may get. Being able to operate with additional mass can be helpful if you know there are more or less patients, and have that flexibility in the supply agreement.
Q There is a great deal of discussion in the field at the moment over the question of whether off-the-shelf or customized raw materials are preferable. What does each option entail for you specifically?
JT: Everyone’s goal is that off-the-shelf products will work. There are some challenges, and for certain processes maybe an off-the-shelf product won’t work, and you will have to go to the customization route. But ideally, off-the-shelf is preferable, and it decreases the amount of time you will need from a development perspective, versus developing a customized solution with a supplier. You can bring it in immediately to your manufacturing processes or QC processes.
One of the key components to that when you explore options with suppliers from a customization perspective, is understanding what that customization really gets you versus what the off-the-shelf product is already capable of doing.
A good example would be if you are doing a harvest of cells during a cell and gene therapy manufacturing process, and your yield is, say, 40%. You need to understanding whether that is really significant. Do you need to look at a solution that increases that yield, or is 40% good enough for what you need to deliver to reach your final products and end goal?
The key is if off-the-shelf works, great – but if not, understand from a customization perspective what the data truly means, and understand whether that is a significant area you need to focus on, or if you should focus on another aspect within your process.
LC: I completely agree. This is going to depend on where you as an organization are with your process, what your goals are, and where you are along the pathway to commercialization.
In an ideal world, an off-the-shelf product is going to be a perfect fit for what you need it to do, will fit well with your strategy for manufacturing, and it is going to be scalable. But as you are moving forward towards when you need to secure that supply chain, there may be a point when you can stop and ask yourself if a customized option will confer an advantage. How do you decide if a custom product is going to be for you? Look at where your process is going, where you want to be, and what you would want of an off-the shelf product to make it fit better.
RL: In an ideal world, off-the-shelf is exactly what you want. Less work goes into determining the specifications and what needs to go into it. But obviously, as I mentioned earlier, we are a growing and a quickly innovating industry. There are times where you have to take some perspective and understand that if the off-the-shelf piece does not fit what you need, you need to pursue a partner that can help you innovate a new, custom approach. And potentially in the future, that can become an off-the-shelf offering for that supplier as well.
If you are looking to improve a step in the process and what you are able to find from an off-the-shelf supplier does not get you to the goal you need, then you go out and speak to custom manufacturers and define exactly what is needed. So it really does depend – both of them have their benefits, but it is dependent on your goal.
LB: Again, it all depends on your process. Can you fit an off-the-shelf material to your process, or is your process in such a place and so unique that you need to have that custom raw material?
Off-the-shelf is nice, but some of these more complex materials are often protected by intellectual property (IP) at the supplier. Understandably, the supplier needs to protect their IP. But when you are trying to set up your own internal specifications for these raw materials, not knowing exactly what the raw material is made of makes it very difficult to set a specification for identity, for example. That is where a custom raw material can come in – then you own the material, you have full knowledge of it, and it is all right there for you going in.
Q Let’s go a little bit deeper into some of the specific pitfalls we touched upon there, that are commonly encountered by the panel with each option. How can they be best avoided?
MB: One of the pitfalls we run into with off-the-shelf GMP products is the time lost, and the risk added in, from the direct handling by the operator. If you are able to close that process with, for example, a customized product in a bag, that can lower the potential of failure there.
Although off-the-shelf GMP can save time, if there is any issue with the processing or handling, or if that product doesn’t exactly fit the workflow, then a custom option may actually be a more cost-effective and timely answer.
For example with products in a bag, if the mass isn’t exactly what is needed to make your media prep, then theoretically, customizing that could save time. It may also be more cost-effective down the line if you don’t need to purchase multiple versions of a product, and things like that.
The pitfalls of customization are upfront timeline and price, but ultimately that might be mitigated with a risk saving.
RL: One of the major pitfalls you have with the off-the-shelf materials is that you will tend to find that with the pace of innovation we are going at, you do not always have what you need to fit into the process. As an example, you find a cell culture medium from an academic paper and it greatly enhances your products, but you are unable to find it as a GMP product off-the-shelf. What you will often do is go and partner with someone to provide you with that material. You cannot get that material off-the-shelf, but being able to convert it to a GMP process will enhance your product greatly.
LB: With custom material the biggest pitfall is obviously the cost and time the user needs to put into it. You sometimes have these biotechnology companies buying each other out, and then they may no longer be able to perform that manufacturing for you.
For off-the-shelf materials, I would say again running into IP issues for the more complex type of materials is the biggest pitfall. There is also a greater risk in terms of your supply chain. There may be other users of that exact same material, and if they suddenly go into a larger scale, from development into commercial, that can mess up your own supply chain. This is where the customization may be better, because you can direct how much you need manufactured each time.
JT: I would add one additional perspective from the business side of things, dovetailing off some of the earlier discussion about single or secondary sourcing.
When you think about going with a customized solution, you are almost creating that single source perspective. You are obviously going to partner with a supplier, so you understand the suppliers capabilities of continuing the supply of that customized solution. If you are investing a significant amount of time and money into a customized solution, you need to understand what their capabilities are.
The other component to that is as you think about a customized solution, how does this all fit into your overall framework of your product portfolio? Are you looking at something as a one-off for one asset you are producing, or a larger area? Look at how you can leverage these off-the-shelf or customized solutions across multiple different assets, as a platform your company can build upon. That has a much more broad impact, even on design of your facilities and things like that.
Q Can you give some further examples of what is involved in taking that off-the-shelf product and fitting it to the client’s process, and converting that to a GMP model?
MB: I view this as two separate examples. One will be changing a small factor of that off-the-shelf product, whether it be the formulation, liquid versus lyophilized, a smaller pack size, or potentially putting it in a bag. Those types of projects tend to be slightly more straightforward, just due to the fact that the specifications may not change from what the off-the-shelf GMP product is. In addition, the process for making it won’t need to change. It is a matter of reconfirming activity, stability, and all of those release criteria, but potentially not a lot of development work is needed.
The other example, which may be a bit more involved, is when you are changing a specification – whether that means starting from scratch with a new protein or a new antibody, or maybe just tightening the endotoxin specifications, and other things along those lines. If you are going to be changing a specification, that is sometimes where it becomes a bit more involved with the process, whether it needs to go to a different lab within the facility, or what the limit of detection is for the assays, and which assays are validated. There are a lot of factors that go into it, depending on whether or not it is effectively a size change, or we are actually changing the specifications of the product itself.
LC: When you are risk assessing your product, it may be that you don’t need GMP yet, but what you have got is not good enough. There is a discussion you may want to have about what the intermediary step is, because when going to a full GMP manufactured product, that timeline may not fit with where you need to be.
We have had several projects, where an intermediate step is good enough for Phase 1, with the end goal in mind that if these programs are taken forwards, then products will in that timeframe be made to GMP, and be appropriate for that next step in the progress of the therapy.
Q When is the key time to partner with a raw material supplier, as far as each of you is concerned, and why?
LB: The simple answer is as early as possible. If you can do it during preclinical development, do it. It takes time to build that relationship with a supplier, and it takes time to get the CDAs in place, and audit and qualify them. It will help you avoid delays later in development.
You can also learn early on whether your supplier is going to be able to fulfil your needs. Let’s say you do need a customized size or container – you need to know that at the beginning. If they cannot deliver that, then you might need to find another supplier. Sometimes it is about shopping around for the correct point of contact within a supplier site. Getting the right people, and developing a relationship there, is the key to being successful.
RL: I would agree with Lili and say the earlier you can do it, the better. As soon as you understand what you want, kick off a sourcing event. As soon as you have the ability to put at least the basic necessities of what you are going to need on paper, you can shoot that out. It is good to be able to start early so you can avoid pitfalls. Additionally, one of the things we find, especially with the pandemic, is that lead times can be much longer than you anticipated. If you wait too long to approach a supplier, it may be too long even for a customer project, because of lead times and especially during the pandemic.
JT: I also agree that when you think about the right time to partner, the sooner the better. One thing I would add to that is the importance of having that continuous and open dialogue with your suppliers as your projects progress and you go through clinical development, and through to commercialization stages. When you think about suppliers, you are probably going to leverage several of them across multiple different assets or programs as well. Understand what lead candidate you are focusing on, what is next within your pipeline, what the timeline is with that, and be upfront with suppliers to ensure they can meet the demand for multiple assets within your overall product portfolio.
Q Mitch – from the supplier side, do you feel that people are engaging with you early enough in their development cycle?
MB: It is ideal when it is as early as possible. I would say it is probably about 50/50 whether someone is coming to us and needs a better product or a change to the product they are using, versus coming to us in the preclinical stage.
One other thing to follow on from what the other panelists were discussing is there is a lot of qualification that goes into picking a vendor. If you are able to collaborate and have a joint experience to optimize that process for whatever reagent it is, you will know those processes are in place to support your other pipeline initiatives as well. It can be a timesaving, once you have that supplier in place, to continuously use them for either off-the-shelf or customization. It can make the process for your other pipeline initiatives simplified as well.
Q Lindsey, what would your key advice be in terms of optimizing these partnerships, as you progress through development towards commercialization?
LC: Everyone has touched on it: talk to us early is definitely the recommendation.
Transparency and communication between manufacturers and suppliers is just so important. All of us on the supplier side want you to succeed; we want you to develop these therapies and have absolute success with them. Part of that is not being afraid to ask us those difficult questions early on. If we are not able to grow with you to supply those needs long term, then we need to be transparent about it. My recommendation is don’t gloss over vague answers. As Mitch alluded to, people are coming to us late in the game and saying “this is where I am at, but this is where I need to be, can you help?” Perhaps its relating to licensing of a particular antibody clone, for example. They might have been better off getting the advice that this is a no-go for what they want to do earlier on. By having that transparency on the supplier side, we can then give that advice and say actually, maybe you would be better off generating an entirely new product to circumvent that.
We cannot emphasize enough that if you are asking those questions early on, and giving us some insight into where you are taking these reagents, we can advise you on what is going to be most appropriate. It might not be what is sitting off-the-shelf, as an RUO raw material – it might be something that you don’t even know we have.
Q It would be great to hear some specifics regarding the issue of scale up. What are the needs of both supplier and end user that need to be met to ensure you can scale up efficiently?
RL: As the industry matures this is going to happen more often than not, but again, it goes back to the ideas of transparency, understanding, and an open dialogue between both the supplier and the company. Saying to the supplier we are looking to approach commercialization, this is the quantity we expect to consume, and making sure they are able to supply that. Working that into your supplier agreement is important as well. Making sure there are contingencies upon that if they have a catastrophic event will potentially allow you to take that to a CMO to get it manufactured.
Open dialogue is probably the most important thing you can have, as well as letting them know you are approaching commercialization and there is going to be additional requirements on that, and potentially information beyond the drug master file that you will need to submit. It is about making sure you have constant communication, especially with your critical supplier, and letting them know what stage you are at and where you plan on taking things.
LB: I agree totally that being very transparent, and providing your forecast as much as you possibly can, will help make sure your supplier won’t overcommit and leave you dangling there. Another thing to consider is ensuring they would not grant exclusivity of a certain raw material or certain target to one supplier over another, leaving you by the wayside.
JT: What it boils down to is having that open and honest transparent communication, and that applies to both parties. From the end user perspective, make sure that what you need is truly communicated to the supplier. From the supplier perspective, make sure you understand those requests or needs from the end user, and be open and honest on whether you think you can meet their expectations.
The other key component to this is as companies engage suppliers or vendors, we are not looking at a one-off solution, we are looking at building a relationship. The crux of that is whether you have that open and honest relationship on both sides.
MB: One of the factors from the supplier side is being able to plan for future need and get those clear forecasts. When you are working with dedicated equipment, and scaling up to potentially multi-gram levels, equipment ordering and procurement can create quite a lead time.
Just as we wouldn’t want to oversell our abilities, we need to make sure we are appropriately managing the project, and have everything in place to pull the trigger when we move past those milestones. It all comes down to communication, along with proper forecasting from both sides.
LC: As a supplier it is ultimately our responsibility to get you what you need, when you need it and where you need it. For that to happen, we need to have transparency on your changing needs and a good forecast is critical to that. Open, clear communication and transparency sums it up.
Reference
Biographies





Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: M Brabec and L Clarke are employees of Bio-techne. R Luke owns stock options as granted as an employee of Adaptimmune, LLC. JP Tomitshen III declares that he has a leadership or fiduciary role in the following boards, societies committees or advocacy groups: BioNJ Manufacturing Advisory Committee Member; ASGCT Commercialization Committee Member; PDA Cell and Gene Therapy Interest Group; ARM Science and Technology Committee; Gene Therapy and Gene-Modified Cell Therapy Committee; Deloitte NextGen Industry Working Group and Accenture Industry Working Group. He is an employee of and has stock options in Legend Biotech USA Inc. The authors declare that they have no other conflicts of interest.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2021 Bio-Techne. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: This article is a transcript of a previously published webinar, which can be found here.
Webinar published: Feb 11 2021; Publication date: Mar 23 2021.
