
Small labels, big challenges: solutions for advanced therapy labeling
Cell & Gene Therapy Insights 2020; 6(8), 1183–1195
10.18609/cgti.2020.128
Accurate and useful labeling of advanced therapies from starting material to final drug product is critical for patient safety, compliance, and treatment delivery. In this article, the authors present label and label printing best practices for advanced therapies and discuss solutions to major challenges faced by sponsors, stakeholders, and regulators. In the context of this article, the terms “label” and “labeling” refer to in-process and final drug labels, not warning labels, package insert content, or other advisories issued and managed by regulatory agencies. As cell/tissue collection and drug product labeling are complex topics, it is also important to note and this article will focus on a few key areas, outlined below. This article will not cover other important labeling topics, including label stock, adhesives, inks, exact label content and layout (other than referring to ISBT128/SEC), label version control, label design, label testing/qualification, or label size and location.
Any Questions?
Today’s advanced therapies are novel and hold great promise, but also pose new challenges that sponsors and stakeholders are rallying to solve. One such challenge is in-process and final product labeling. The requirement for appropriate and approved labeling is not new in biotech and pharma – it is foundational to every drug product and was recently reinforced by the FDA’s guidance for gene therapy INDs [1]U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/download. Advanced therapies, however, introduce patient or donor cellular raw material into the supply chain, resulting in a much more complex process and treatment journey to produce these therapies (Figure 1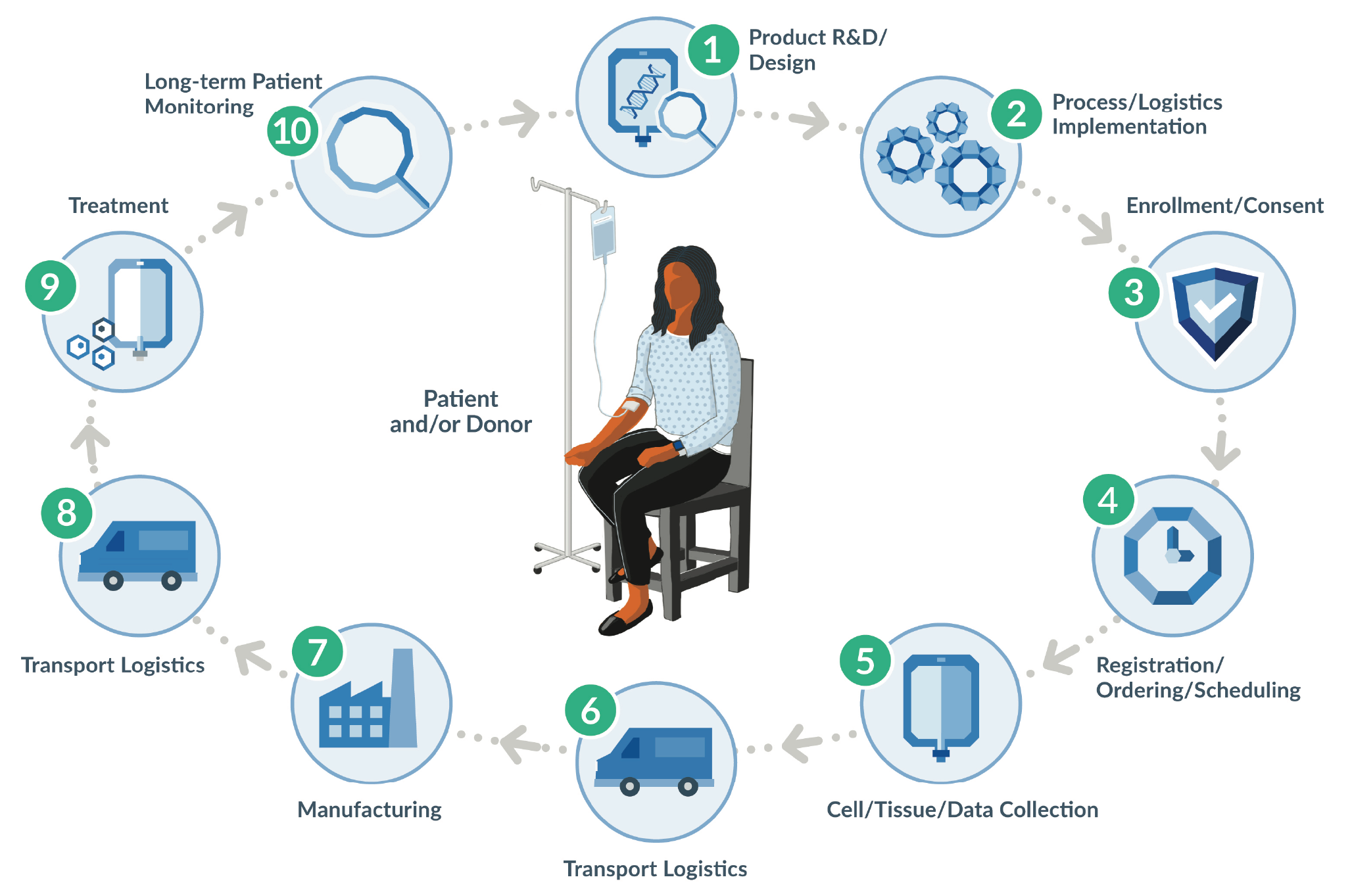
As a critical patient-product identifier and safety measure, advanced therapy drug product labels must often be created for each individual patient and built in real time, with important and variable information added to the label at key steps in the cell collection and manufacturing process. Healthcare Providers (HCPs), in performing cell collections, now find themselves collecting starting material and are required to comply with cGMP information gathering and labeling processes. Given the patient-specific, real-time nature of advanced therapy labeling, labels can be pre-printed and shipped to clinical sites, which simplifies some aspects of label creation but can also result in storage, matching, and disposal problems. Or manufacturers can choose to have HCPs print labels at the clinical site, which reduces storage and matching problems but can cause challenges related to hardware, hospital IT compatibility, and printing equipment.
With more than 1,060 advanced therapy drug products and clinical trials operating at medical centers world-wide each with a different process and set of Standard Operating Procedures (SOPs), the need for simplified, standardized labeling processes is acute [2]Alliance for Regenerative Medicine. ARM Annual Report & Sector Year in Review: 2019. Washington, D.C. Alliance for Regenerative Medicine. 2020; 30: https://alliancerm.org/publication/2019-annual-report/. The catalyst for labeling standardization began with a public health issue related to unsafe handling of donated blood in the 1990s, and the subsequent discovery that traceback capabilities did not exist. The FDA stepped in and initiated what we now consider standard traceability of blood and tissue donation products. Building on this foundation, the International Council for Commonality in Blood Banking Automation (ICCBBA) published the ISBT 128 global standards in 1994 [3]International Council for Commonality in Blood Banking Automation (ICCBBA) ©2020: https://www.iccbba.org/isbt-128-basics/get-to-know-isbt-128. Cell therapy pioneers adopted these baseline labeling standards for human blood and tissue products at the FDA’s direction. This initially served the industry well – to a point – and raised the bar on patient safety.
However, as more advanced therapies enter the clinic and approach commercial approval, it has become clear that there is a gap in labeling standards when implemented for today’s advanced therapies. The ISBT 128 and SEC standards are in effect for blood and tissue collection or donation activities, but the standards were not established with the expanded and complex advanced therapy supply chain in mind, where traceability from order through manufacturing, treatment and beyond is required [4]International Council for Commonality in Blood Banking Automation (ICCBBA). Introductory Booklets ©2020: https://www.iccbba.org/isbt-128-basics/introductory-bookletsInternational Council for Commonality in Blood Banking Automation (ICCBBA). Introductory Booklets ©2020: https://www.iccbba.org/isbt-128-basics/introductory-booklets, [5]International Council for Commonality in Blood Banking Automation (ICCBBA). ISBT 128 Standard: Labeling of Cellular Therapy Products: Version 1.2.0. San Bernardino, CA. November 2018: https://www.iccbba.org/uploads/6c/db/6cdba776ce2ba38a0c21776f2b7ee95b/ST-004-ISBT-128-Standard-Labeling-of-Cellular-Therapy-Products-v1.2.0.pdfInternational Council for Commonality in Blood Banking Automation (ICCBBA). ISBT 128 Standard: Labeling of Cellular Therapy Products: Version 1.2.0. San Bernardino, CA. November 2018: https://www.iccbba.org/uploads/6c/db/6cdba776ce2ba38a0c21776f2b7ee95b/ST-004-ISBT-128-Standard-Labeling-of-Cellular-Therapy-Products-v1.2.0.pdf, [6]European Commission. Overview, standards documents and implementation guides. © European Union, 1995-2020: https://ec.europa.eu/health/blood_tissues_organs/tissues/single_european_code_en#:~:text=The%20%22Single%20European%20Code%E2%80%9D%20or,type%20of%20tissue%20or%20cells.European Commission. Overview, standards documents and implementation guides. © European Union, 1995-2020: https://ec.europa.eu/health/blood_tissues_organs/tissues/single_european_code_en#:~:text=The%20%22Single%20European%20Code%E2%80%9D%20or,type%20of%20tissue%20or%20cells.. As a result, a cross-industry standards updating effort is now underway, which will be discussed later in this article.
Label issues are a major reason for the FDA rejecting or questioning New Drug Applications (NDA) and Biologics License Applications (BLA) [7]US Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, Office of New Drugs (OND). Manual of Policies and Procedures, Good Review Practice: Refuse to File. Silver Spring, MD. The FDA. September 5, 2018; 15, 17: https://www.fda.gov/media/87035/download. Revisiting proven best practices related to donor material and drug product labeling will help sponsors and stakeholders bridge the gap between existing standards and emerging standards, and enable manufacturers to bring life-saving advanced therapies to patients in need more safely and efficiently.
Seven proven practices
Experience demonstrates that certain practices lead to successful labeling and support the ability to deliver safe treatments to patients. The following top seven labeling practices are proven to work across multiple therapies, for both clinical and commercial phase products:
- Standardized formats
- Patient privacy and patient identifiers
- Complete Chain of Identity (COI) and Chain of Custody (COC)
- Multi-language capabilities
- On-demand label printing
- Printing to any existing and approved printer
- Collaboration with regulatory agencies
Standardized formats
Standardization across materials, products, and processes decreases risk and errors while increasing efficiency and scalability. This is foundational to cGMP and especially important in a distributed ecosystem such as the advanced therapy supply chain, where many partners are not accustomed to cGMP practices. When established standards exist, utilizing them is a key to compliance. The FDA has certain baseline requirements for labeling (see Figure 2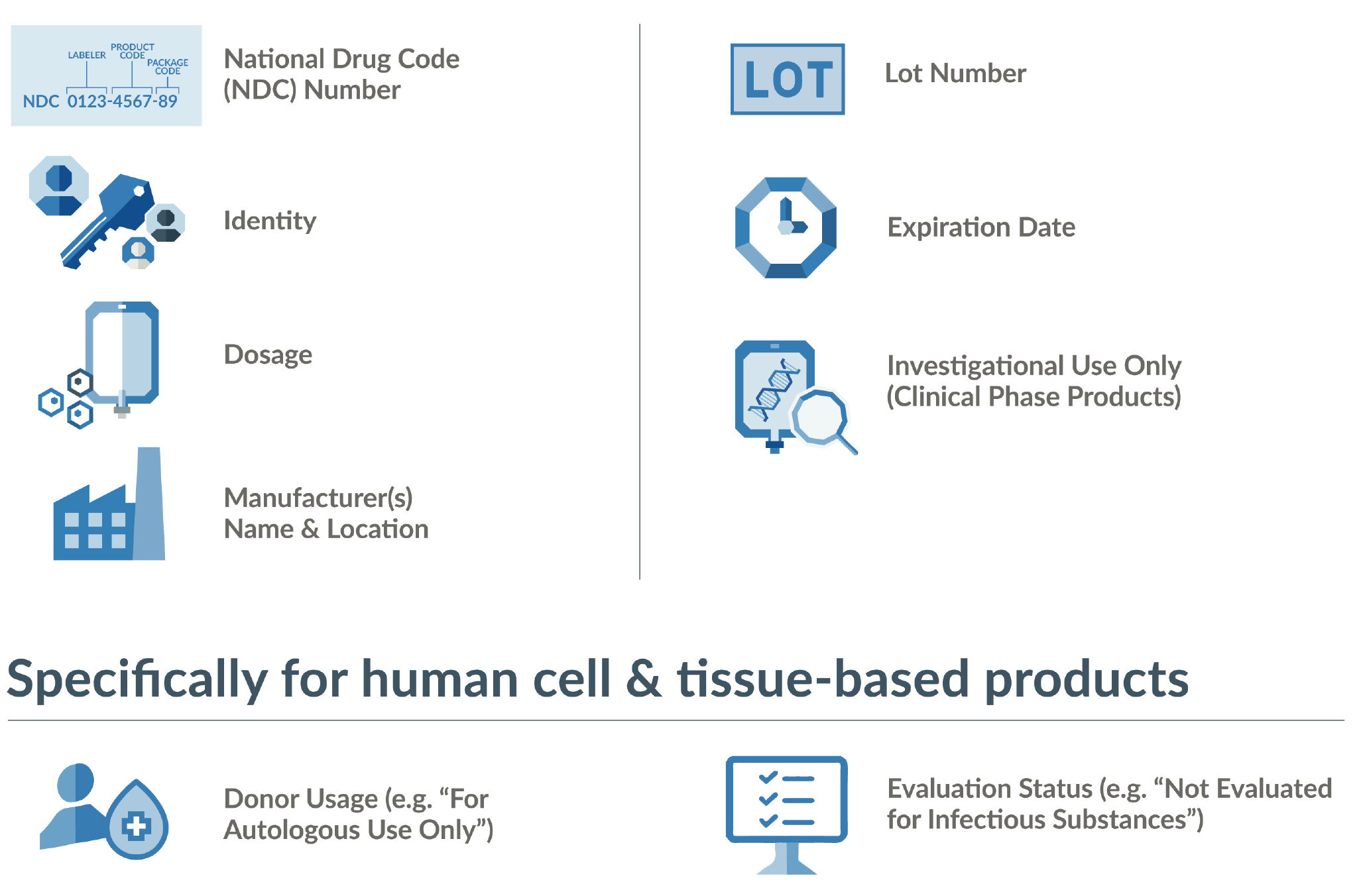
An additional consideration is the importance of providing label information via scannable barcodes and human readable formats. Space is at a premium on labels, and in this digital age, it is tempting to count on reading barcodes with scanners. But maintaining continuity and integrity of patient identifiers across the entire supply chain is paramount for avoiding product mix-ups. That may mean having the ability to verify information without the benefit of a digital tool.
The independent, non-profit Standards Coordinating Body is facilitating an industry-wide working group to update the ISBT 128 standards for apheresis collection labels and establish minimum label requirements for apheresis cell collections (Figure 3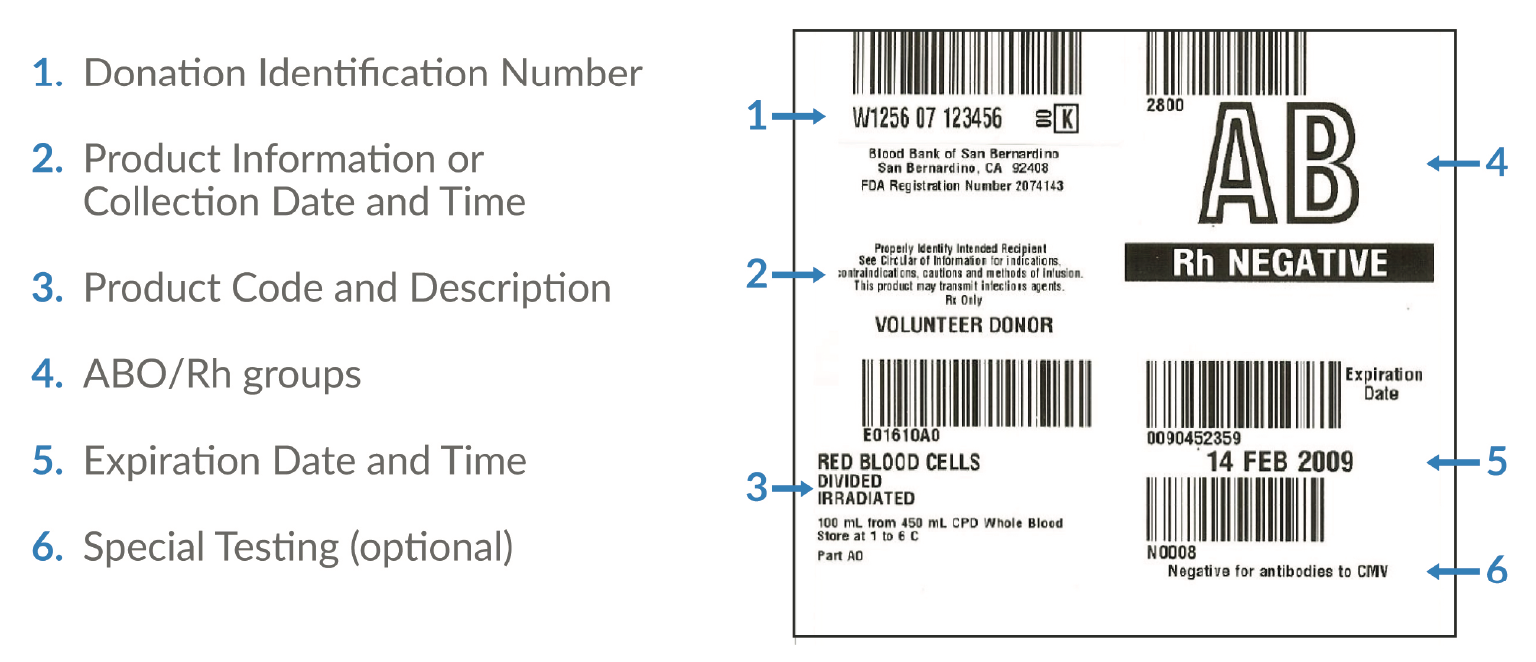

Patient privacy & patient identifiers
As a critical support for patient safety, labels must contain some patient-identifying information. The nature of that information, such as name, initials, or date of birth, is a frequent topic of debate. Alongside patient safety, patient privacy is also important.
Amid privacy concerns, it’s important to keep in mind that name, initials, and date of birth are typical patient identifiers used in medical centers and for prescriptions to ensure that the right product is administered to the right patient. This is part of standard practice on drug product packaging, and HCPs are trained to use this information appropriately. This practice is demonstrated in the use of patient information on approved cell therapy product labels such as Provenge® [14]Provenge® product label: https://www.drugs.com/pro/provenge.html, Yescarta® [15]Yescarta® product label: https://www.accessdata.fda.gov/spl/data/783b1236-8e53-41a7-922a-08bdfb7517ef/783b1236-8e53-41a7-922a-08bdfb7517ef.xml, Kymriah® [16]Kymriah® product label: https://www.accessdata.fda.gov/spl/data/32b00de7-ce25-48db-abce-cfe0d65dbcc0/32b00de7-ce25-48db-abce-cfe0d65dbcc0.xml, and Zynteglo® [17]Zynteglo® product label: https://www.ema.europa.eu/en/documents/product-information/zynteglo-epar-product-information_en.pdf (EMA approved).
The European Union’s (EU) General Data Protection Regulation (GDPR) has been reshaping the way data is handled across every industry sector, including clinical research, by strengthening and standardizing the protection of personal data. Navigating GDPR and traceability requirements in the EU may require further expert help and collaboration with regulators – a best practice discussed in more detail later .
Concerns over visibility of patient information on external secondary and tertiary packaging (e.g. shipping labels) that is more broadly viewable can be addressed with anonymized information. Additional persistent, transparent patient identifiers can be created and tracked in ways that are consistent with patient privacy regulations. One such set of identifiers is commonly referred to as Chain of Identity, or COI, which we’ll discuss next.
Chain of Identity (COI) & Chain of Custody (COC)
COI and COC are the cornerstone of three important success factors for any drug product – patient safety, regulatory compliance, and operational efficiency (Figure 5
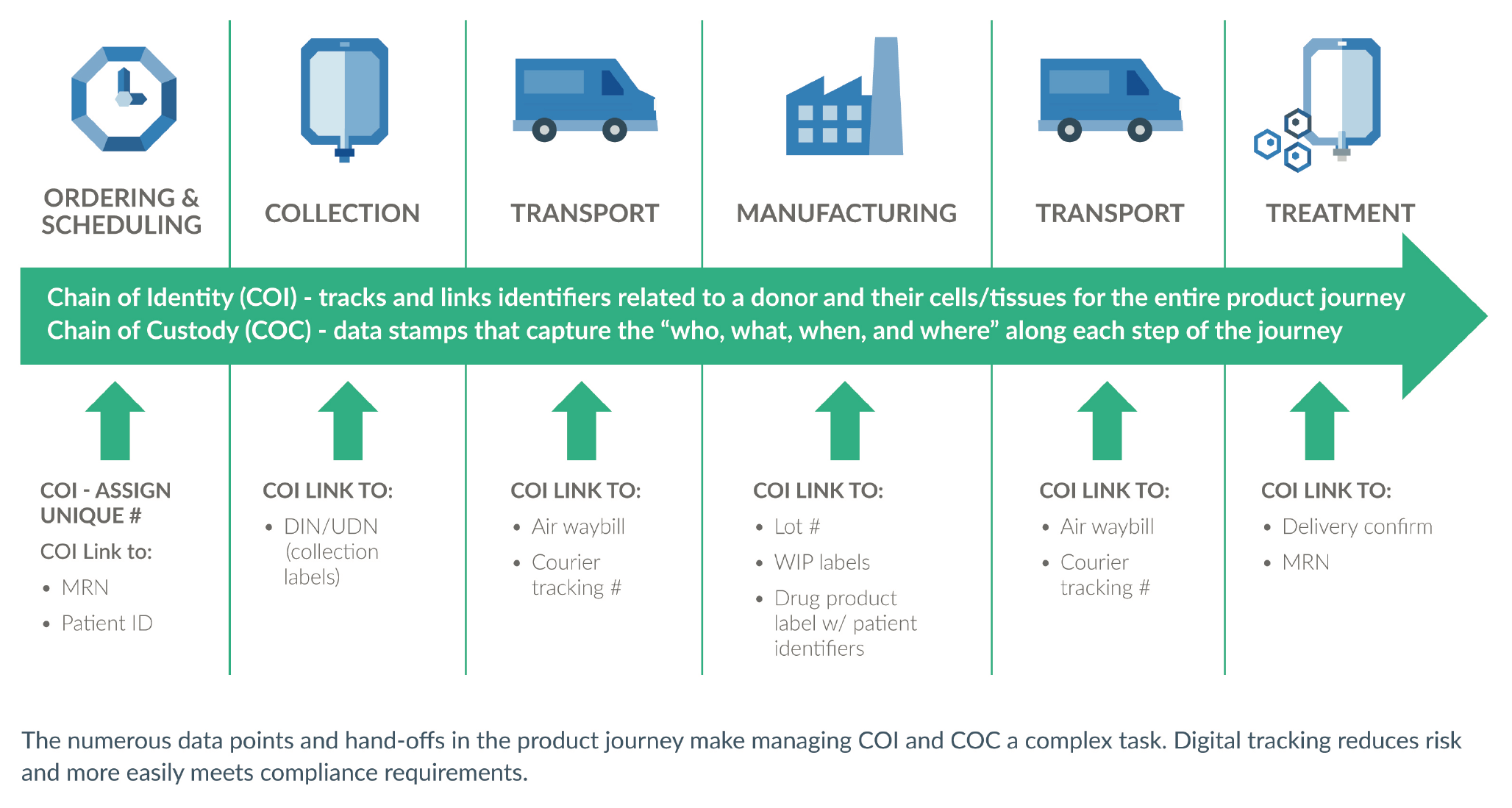
The established best practice for advanced therapy COI and COC tracking is the use of a digital system. The numerous touch points and physical locations in the product journey make manual tracking time-consuming and error-prone. It is too risky to leave patient safety to chance in this way. Additionally, routine compliance becomes onerous, and product approval may be a challenge. Regulatory reviewers want an application to demonstrate that traceability is well in hand (Figure 7
Multi-language capabilities
The complex, distributed ecosystem of advanced therapies often means supply chain partners and patients are located in many geographic regions, both within the United States and across the globe. The need for starting material and drug product to cross borders, or to be produced and delivered locally in a region different from that of the biopharma company, presents additional labeling challenges. Labels must be in compliance with local, national and international requirements, depending on the situation, and must also be relevant and useful to those handling and processing the material or product. One important aspect of this is the ability to generate raw material, in-process, and final product labels in multiple languages.
Ensuring patient safety means that personnel at local care sites and other supply chain partners must be able to easily and definitively read key pieces of information on labels, meaning the information must be presented in the appropriate language(s). This is challenging due to the limited physical space available on many labels, the sheer number of possible languages and language formats a company may need to support (left to right, right to left, top to bottom), and the variability in translations. Working with experienced partners and regulators on label design, formatting, and printing helps ensure that the essential information is included, presented in only the correct languages required for that geography, and that the physical space on the label is used efficiently.
Solutions such as Vineti’s Personalized Therapy Management Platform (PTM; Vineti solution) [19]Vineti’s Personalized Therapy Management Platform: https://vineti.com/solution/featuresVineti’s Personalized Therapy Management Platform: https://vineti.com/solution/features come equipped and ready to support multi-language labels (including character based languages), providing the ability to deploy and maintain standardized, compliant labeling across multiple geographies for local use. Additionally, establishing or procuring approved and standardized phrase libraries ensures accurate, consistent translations every time and cuts significant time out of the process for creating, reviewing, and revising labels. The benefits of proactively tackling multi-geography labeling challenges pay off early, even if there are only a few regions involved. As clinical trials progress and expand, and a therapy moves toward commercialization, it can be difficult to scale one-off or manual processes. Having established multilingual capabilities early on to scale up and out across geographies will save time and money getting therapies approved and to the patients who need them most.
On-demand label printing (vs print & ship)
One of the decisions that advanced therapy manufacturers face is whether to print labels in advance and ship them to the partner sites, or establish the capabilities for partner sites to print labels on-demand, prior to, or during, processing. This seemingly small decision has significant factors to consider, and is a major consideration as therapy delivery scales. For many reasons, on-demand label printing is the recommended and most compliant option.
Most importantly, patient safety is easier to ensure with on-demand label printing. Patient ID verification is linked to label printing and the patient is paired with their labels from the outset. On-demand is also much simpler to manage from a compliance and supply chain standpoint. Label reconciliation is a key cGMP compliance activity to prevent product mislabeling (or product mix-ups) [20]Current Good Manufacturing Practice for Finished Pharmaceuticals: Packaging and Label Control – Labeling Issuance, 21 C.F.R. Part 211 Subpart G Sect. 211.125 (2019). . Every label must be accounted for, and pre-printed labels create additional touch points and opportunities for labels to be lost and unverified. Operationally, pre-printed labels create a whole new ‘supply chain’ that must be managed, requiring additional and costly resources, and putting additional and unnecessary pressure on an already time sensitive process.
Printing to existing printers
There have been different approaches and challenges related to label printers as well. One option was to deploy dedicated printers for each therapy to partner sites. This may be problematic in some cases, because additional complexity and cost is unnecessarily introduced into the supply chain, there are additional security considerations, and scalability is hampered. Ecosystem partners such as HCPs and clinical sites may have limited space for additional hardware, and deploying dedicated printers to individual partner sites involves installation, training, logistics, and maintenance requirements that are time-consuming, personnel intensive, and expensive.
A more sustainable and scalable model is to utilize existing printers at partner sites for label printing. This is time and money saving for both the partner and the drug developer and simplifies one aspect of a complicated process. It is also more secure to utilize hardware already in operation within a site’s IT system, and modern cloud-based solutions provide enhanced security and remote management. This ability is a feature of Vineti’s Personalized Therapy Management platform (PTM), which provides a turnkey solution for label printing (for more information on PTM, please see [19]Vineti’s Personalized Therapy Management Platform: https://vineti.com/solution/featuresVineti’s Personalized Therapy Management Platform: https://vineti.com/solution/features).
Collaboration with regulatory agencies
The FDA’s mission is “…protecting the public health by assuring the safety, efficacy and security of human and veterinary drugs, biological products, medical devices…” (Figure 8
One important way the agency carries out this mission is through the requiring and approving of in-process and final product labeling. It is worth mentioning again that label issues are cited by FDA regulators as a major reason for rejecting or questioning New Drug Applications (NDA) and Biologics License Applications (BLA).
Frequent collaboration with regulatory agencies cannot be stressed enough. This helps ensure filing acceptance and avoids the many potential pitfalls for advanced therapy labels. Beginning with IND filing, regulatory guidance puts emphasis on the importance of labeling, further indicating that early focus and collaboration on labeling is smart [1]U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/downloadU.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/download. If left until late in the process, labeling processes and details may present an unexpectedly time-intensive, complex issue to solve. This holds in the United States, and is also important globally, where there may be multiple agencies involved and differences from region to region. Each drug product will have something unique that will need to be addressed specifically by regulators. It is better to ask questions up front, rather than to wait until the application is filed. Early conversations and feedback during product development can eliminate clinical holds and costly, timeline-breaking re-work – in addition to delays in getting treatment to critically ill patients.
Translational insights: standards for scale
As advanced therapies grow in number and reach, sponsors and stakeholders are developing standards to enable a patient-centric drug product ecosystem. The Standards Coordinating Body (SCB) is conducting FDA-funded work to carry out standards directives in the 21st Century Cures Act, Section 3036. As part of its work on advanced therapy standards, the SCB, in partnership with ICCBBA, is building on the existing labeling standards for cell collection products and modifying them to accommodate the current state – for autologous and allogeneic products – to help prepare the industry for future success [22]Food and Drug Administration. Standards Development for Regenerative Medicine Therapies, Section 3036 of the 21st Century Cures Act. Contents not copyrighted: https://www.fda.gov/vaccines-blood-biologics/standards-development-regenerative-medicine-therapies https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act.
This SCB industry working group on labeling is composed of a variety of ecosystem stakeholders and subject matter experts who can provide the perspective and experience needed to develop suitable standards [23]Standards Coordinating Body (SCB). Project: Base Labeling Requirements for Apheresis Products for Regenerative Medicine Manufacturing. Copyright © 2020: https://www.standardscoordinatingbody.org/project-base-labeling-apheresis-products. Some of the standards being evaluated in relation to apheresis collection labels include a working common definition of COI and COC, label content, layout, required data, and data formats. Timelines for gathering final input and publishing the updated standards are being determined in collaboration with ICCBBA. To see the draft standards document, please visit the ICCBBA website [24]ICCBBA, Documents for Review: https://www.iccbba.org/tech-library/iccbba-documents/documents-for-review. To learn more about this important effort and get involved, please contact SCB [25]SCB, Base Labeling Requirements for Apheresis Products for Regenerative Medicine Manufacturing: https://www.standardscoordinatingbody.org/project-base-labeling-apheresis-products.
Looking to the future, such standards will become more important than ever. The number of advanced therapies in the R&D pipeline is increasing, with a goal of making more therapies safer and capable of being delivered on an out-patient basis. Scaling will involve expansion more broadly into community-based settings. This accessibility across all types of medical centers is critical for patients. Therefore, supporting the ability for smaller, more distributed clinical sites to collect starting material, deliver treatments, and manage patient-specific labels is required for greater access. Simple, flexible, compliant label printing is one important piece of this model. By following proven practices and working together to develop and implement standards, advanced therapy sponsors and stakeholders will take a critical step towards a future of greater advanced therapy access for all.
Heidi Hagen has been an Operations Executive in the Biotechnology industry for 30 years and is a Co-founder of Vineti. She has overseen the delivery of more than 100,000 doses of personalized cell therapy and has guided numerous therapies through clinical development on to commercialization. She has an extensive and proven track record in leading operations and commercializing innovative technologies ranging from recombinant protein/device combinations to the first active immune cell therapy, Provenge®. Heidi is on the Board of Directors for Vericel Inc., Ziopharm Inc., and Lykan Biosciences. Previously, Heidi was the Global COO for SOTIO, in Prague, Czech Republic with a US office in Boston, MA. Before joining SOTIO, she worked for Dendreon for ten years as Senior Vice President of Operations, and 10 years with Immunex Corporation in a range of roles in drug development and operations management. Heidi has a BS in Cell and Molecular Biology, an MS in Bioengineering, and an MBA from the University of Washington.
Christophe Suchet brings more than 20 years of information technology and senior biopharmaceutical IT experience to Vineti. Most recently, he served as Vice President of IT for Kite Pharma, Inc., where he developed the essential technology systems that helped Kite’s first cell therapy receive FDA approval, scale rapidly, and secure the landmark sale of the business to Gilead Sciences, Inc. Prior to Kite Pharma, Christophe served as Vice President of IT at Pharmacyclics Inc., a clinical stage and commercial biopharmaceutical company that grew in two years from no product revenue, to $1 billion revenue, to acquisition by AbbVie for $21 billion. Christophe has served as Genentech’s Director, IT Pharma Development Applications, and as IT Director, SAP Center of Excellence & Enterprise Applications. He received his MS in Biology and Economy from AgroParisTech.


References
1. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs): Guidance for Industry. Silver Spring, MD. The FDA. January 2020; 4,16–17: https://www.fda.gov/media/113760/download Crossref
2. Alliance for Regenerative Medicine. ARM Annual Report & Sector Year in Review: 2019. Washington, D.C. Alliance for Regenerative Medicine. 2020; 30: https://alliancerm.org/publication/2019-annual-report/ Crossref
3. International Council for Commonality in Blood Banking Automation (ICCBBA) ©2020: https://www.iccbba.org/isbt-128-basics/get-to-know-isbt-128 Crossref
4. International Council for Commonality in Blood Banking Automation (ICCBBA). Introductory Booklets ©2020: https://www.iccbba.org/isbt-128-basics/introductory-booklets Crossref
5. International Council for Commonality in Blood Banking Automation (ICCBBA). ISBT 128 Standard: Labeling of Cellular Therapy Products: Version 1.2.0. San Bernardino, CA. November 2018: https://www.iccbba.org/uploads/6c/db/6cdba776ce2ba38a0c21776f2b7ee95b/ST-004-ISBT-128-Standard-Labeling-of-Cellular-Therapy-Products-v1.2.0.pdf Crossref
6. European Commission. Overview, standards documents and implementation guides. © European Union, 1995-2020: https://ec.europa.eu/health/blood_tissues_organs/tissues/single_european_code_en#:~:text=The%20%22Single%20European%20Code%E2%80%9D%20or,type%20of%20tissue%20or%20cells. Crossref
7. US Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research, Office of New Drugs (OND). Manual of Policies and Procedures, Good Review Practice: Refuse to File. Silver Spring, MD. The FDA. September 5, 2018; 15, 17: https://www.fda.gov/media/87035/download Crossref
8. Current Good Manufacturing Practice for Finished Pharmaceuticals: Packaging and Label Control, 21 C.F.R. Part 211 Subparts G and J (2019): https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211&showFR=1&subpartNode=21:4.0.1.1.11.7https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211&showFR=1&subpartNode=21:4.0.1.1.11.10 Crossref
9. Drugs: General – Labeling, 21 C.F.R. Subpart A Sect. 201.2 (2019): https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=201.2 Crossref
10. General Biological Products Standards – Labeling Standards, 21 C.F.R. Part 610, Subpart G (2019): https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=610&showFR=1&subpartNode=21:7.0.1.1.5.7 Crossref
11. Drugs: General – Labeling, 21 C.F.R. Part 201 Subpart B Sect. 201.5 (2019): https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=201&showFR=1&subpartNode=21:4.0.1.1.2.2 Crossref
12. Regulations Under Certain Other Acts Administered by the Food and Drug Administration – Human Cells, Tissues, and Cellular and Tissue-based Products, 21 C.F.R. Part 1271.9 (2019): https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=1271.90 Crossref
13. Standards Coordinating Body (SCB). Copyright © 2020: https://www.standardscoordinatingbody.org/ Crossref
14. Provenge® product label: https://www.drugs.com/pro/provenge.html Crossref
15. Yescarta® product label: https://www.accessdata.fda.gov/spl/data/783b1236-8e53-41a7-922a-08bdfb7517ef/783b1236-8e53-41a7-922a-08bdfb7517ef.xml Crossref
16. Kymriah® product label: https://www.accessdata.fda.gov/spl/data/32b00de7-ce25-48db-abce-cfe0d65dbcc0/32b00de7-ce25-48db-abce-cfe0d65dbcc0.xml Crossref
17. Zynteglo® product label: https://www.ema.europa.eu/en/documents/product-information/zynteglo-epar-product-information_en.pdf Crossref
18. European Commission, EudraLex. The Rules Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice: Guidelines on Good Manufacturing Practice Specific to Advanced Therapy Medicinal Products. London, UK. The European Commission. November 22, 2017; 8, 10, 30: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf Crossref
19. Vineti’s Personalized Therapy Management Platform: https://vineti.com/solution/features Crossref
20. Current Good Manufacturing Practice for Finished Pharmaceuticals: Packaging and Label Control – Labeling Issuance, 21 C.F.R. Part 211 Subpart G Sect. 211.125 (2019). Crossref
21. Food and Drug Administration. FDA Mission. Contents not copyrighted: https://www.fda.gov/about-fda/what-we-do#mission Crossref
22. Food and Drug Administration. Standards Development for Regenerative Medicine Therapies, Section 3036 of the 21st Century Cures Act. Contents not copyrighted: https://www.fda.gov/vaccines-blood-biologics/standards-development-regenerative-medicine-therapies https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act Crossref
23. Standards Coordinating Body (SCB). Project: Base Labeling Requirements for Apheresis Products for Regenerative Medicine Manufacturing. Copyright © 2020: https://www.standardscoordinatingbody.org/project-base-labeling-apheresis-products Crossref
24. ICCBBA, Documents for Review: https://www.iccbba.org/tech-library/iccbba-documents/documents-for-review Crossref
25. SCB, Base Labeling Requirements for Apheresis Products for Regenerative Medicine Manufacturing: https://www.standardscoordinatingbody.org/project-base-labeling-apheresis-products Crossref
Affiliations
Heidi Hagen
Co-founder and Advisor, Vineti, Inc., 633 Howard Street, San Francisco, CA, USA
Christophe Suchet
Author for correspondence:
Chief Product and Compliance Officer, Vineti, Inc., 633 Howard Street, San Francisco, CA, USA
info@vineti.com

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors declare that they have no conflicts of interest.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2020 Hagen H & Suchet C. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited.
Revised manuscript received: Oct 10 2020; Publication date: Oct 21 2020.