Recent advances in non-viral vectors for gene therapy & vaccination
Cell Gene Therapy Insights 2017; 3(2), 95-101.
10.18609/cgti.2017.012



While the majority of gene therapy vectors are derived from viruses, their use introduces the issues of immunogenicity, carcinogenicity, insertional mutagenesis and reversion to replication competency (infectivity). In addition, viral vectors often come with manufacturing bottlenecks such as scalability and processing. In order to avoid the problems associated with viral vectors, investigators turn to the use of naked DNA, usually in the form of non-integrating bacterial plasmids, which have now (as of February 2017) been used in 427 clinical trials [1,2]. So far, there are no regulatory agency approved drugs for humans that are based on plasmids, although four veterinary vaccines have been approved for use in animals [3]. The primary difficulties of using plasmids are: delivery into cells; subcellular trafficking (including nuclear access); and obtaining adequate expression over time, especially in dividing cells where the episomal non-replicative plasmid is diluted at least two-fold in each cell division cycle. In addition, it would be desirable to have robust plasmid manufacturing at a commercial scale.
Delivery into cells can now be obtained by a number of methods, including chemical transfection reagents, electroporation, sonoporation, nanoparticles, hydrodynamic delivery, microneedles, naked DNA, nucleofection, gene gun, jet injection and so on [4–6].
In order for transcription of foreign genes to occur, the plasmid must gain access not only to the cytoplasm (by direct entry or by endocytosis), but also to the nucleus, usually through nuclear membrane disruption during mitosis, or alternatively, by trafficking through nuclear pore complexes (NPCs) into quiescent cells (Figure 1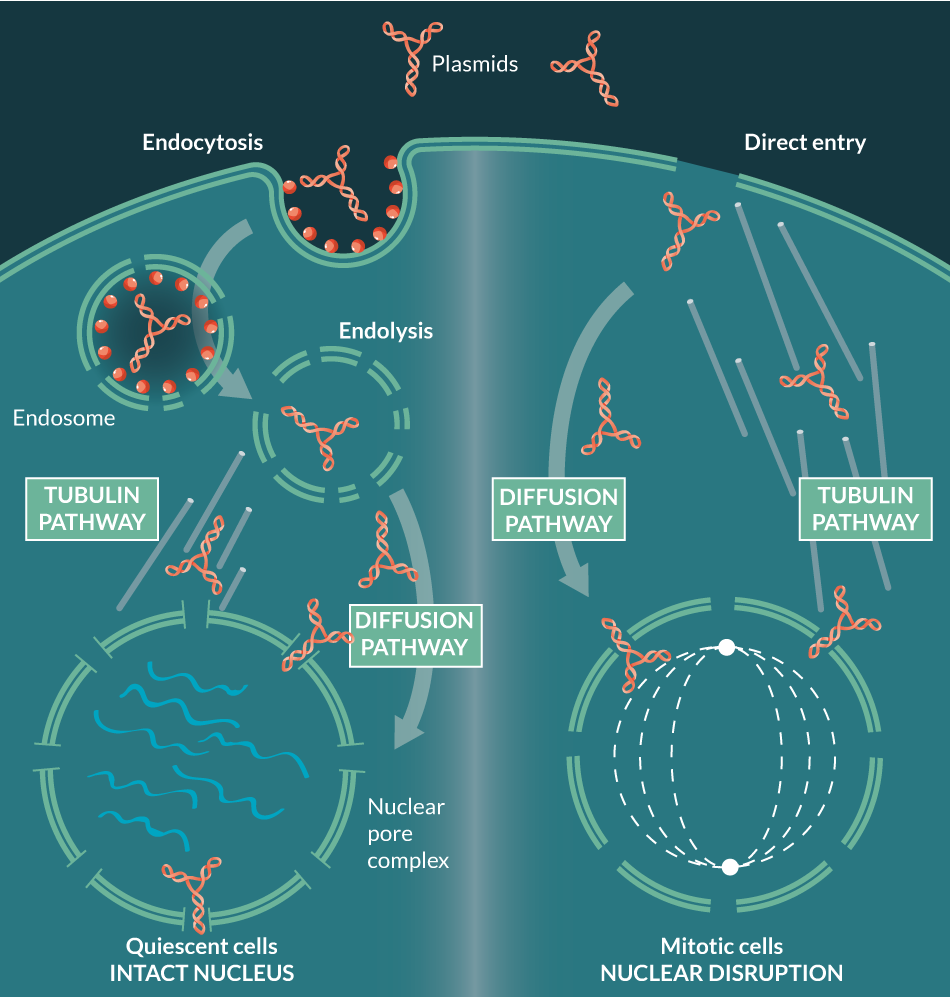
In order to attain improved expression over time, it is important to have highly optimized transgenes and to have bacterial vector sequences that are compatible (preferably synergistic) with expression in eukaryotic cells. In addition, it has recently been shown that plasmid gene silencing is primarily caused by having approximately >1kb of bacterial DNA in the vector backbone [9]. First-generation DNA plasmids have large >2 kb bacterial backbones comprised of the pUC replication origin, an antibiotic resistance marker and extraneous bacterial DNA sequences (Figure 2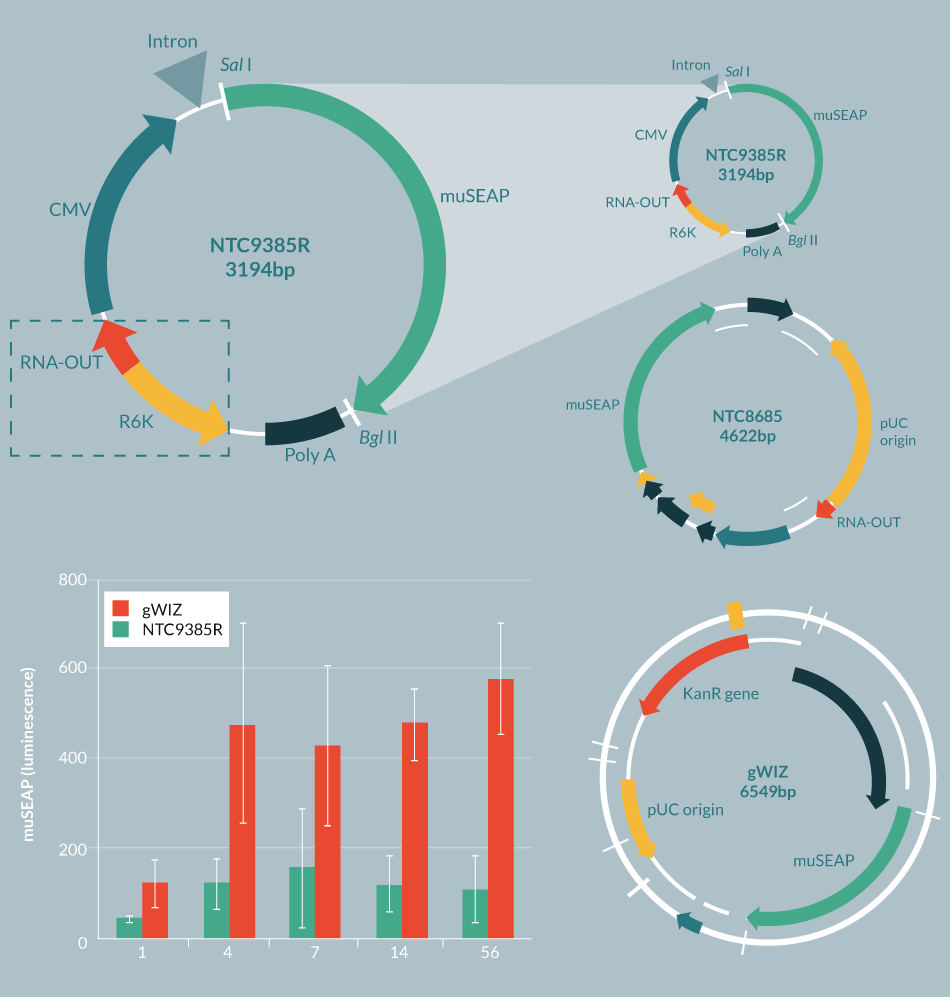
The failure of first-generation DNA plasmids to perform adequately in many early clinical trials has led investigators to design and make vectors with improved efficacy, safety and industrial production [11]. Nature Technology Corporation (NTC) has spent the past 15 years developing innovations aimed at solving technical issues related to the use of DNA plasmids. The initial goals were to remove all non-essential sequences from the plasmid backbone (including extraneous bacterial DNA flanking the selection marker and replication origin, Z DNA, cruciform, palindromes, repeats, cryptic splice sites, RNA secondary structures, human genome homology, alternative reading frames, cryptic promoters or chi sites) and to provide a useful variety of subcellular targeting tags and transcriptional promoters. This resulted in the pDNAVACC™ family of vectors, intended for use as DNA vaccines and for gene therapy [12].
Next, antibiotic resistance genes were replaced with a small RNA selectable marker (RNA-OUT) [13], which allows selection with sucrose rather than antibiotics, and which has the serendipitous effect of dramatically improving gene expression from the eukaryotic side of the vector [14,15]. This resulted in the 1.7 kb bacterial backbone RNA-OUT pUC origin selection-replication origin NTC ‘8’ series of vectors (Figure 2) [14,16]. These vectors are currently in a number of Phase 1 and 2 clinical trials with no serious adverse events, have shown positive efficacy ex vivo and in vivo [17–19], and have been licensed by biopharmaceutical/drug development companies.
Next, plasmids with R6K or ColE2 miniature origins of replication (mini-ori) were devised, which use engineered E. coli host strains that allow conditional replication of plasmids only in the engineered strains (also helpful to prevent propagation of the plasmids in the environment, which has been found to be a problem with traditional pUC-derived plasmids [20]). These new mini-vectors, called Nanoplasmids™ [21,22] are among the smallest plasmid bacterial replication and selection backbones (<0.5 kb, including mini-ori and RNA-OUT). They have expression levels higher than the NTC 8 series vectors, are resistant to gene silencing and do not require further processing/recombination in order to maintain high levels of expression (Figure 2). The 1.7 kb NTC ‘8’ series RNA-OUT, pUC origin cassette was also used by the Kay Lab at Stanford University to create mini-intronic plasmid (MIP) vectors [15], which resemble Nanoplasmids™, but contain the RNA-OUT, pUC origin cassette within an intron, rather than within the spacer region (located between the poly(A) site and the enhancer of the eukaryotic transcription unit sequences). MIP vectors, like Nanoplasmid™ vectors, increase transgene expression up to ten-fold compared with plasmid or minicircle vectors and alleviate transgene silencing [15]. The RNA-OUT mini-ori cassettes when substituted for the RNA-OUT pUC selection-replication cassettes in MIP vectors show similarly improved vector performance [21]. NTC’s Nanoplasmids™ have been tested thoroughly by many companies and labs throughout the world, but have not advanced to human trials yet. Nevertheless, each of the elements of Nanoplasmids™ (RNA-OUT, R6K) have separately been tested in the clinic without problems.
The optimized minimal antibiotic-free plasmid backbones have utility beyond improving performance of non-integrating bacterial plasmids, and may be useful in applications where minimal vector backbone minicircle vectors have demonstrated improved performance. For example, Sleeping Beauty Transposon minicircle vectors are more effective and less toxic than canonical plasmid backbones for engineering CAR T-cells [23]. Minicircle versions of episomally maintained S/MAR non-viral DNA vectors show improved expression and replicative establishment frequency compared to plasmids [24,25]. It has also been demonstrated that antibiotic resistance markers adjacent to adeno-associated viral (AAV) inverted terminal repeats (ITRs) are often packaged into viral particles during production [26]. Antibiotic-free backbones, along with minicircle vectors [26], have application to eliminate antibiotic marker contamination of AAV particles. Of relevance to this application, the <0.5 kb antibiotic free RNA-OUT mini-ori selection-replication cassette, when positioned within an AAV vector intron, improved in vivo AAV-mediated transgene expression 40–100-fold over that obtained with a conventional AAV vector [27]. This suggests that elimination of antibiotic marker contamination of AAV viral particles using an intronic mini-ori selection-replication cassette may additionally improve AAV vector performance.
Finally, for industrial/commercial production of plasmids, NTC has developed a plasmid cell banking and fermentation process (HyperGRO™) [28–31] that results in dramatically improved yields (1–3.5 g/L) of plasmid and Nanoplasmid™ DNA, compared to traditional methods (typically 100–500 mg/L). The fermentation involves a batch growth phase in semi-defined media, followed by nutrient limited exponential growth (µ = 0.12 h-1) at reduced temperature (30 °C), followed by a mid-exponential phase temperature shift (42 °C) to induce plasmid hyper-proliferation. The fermentation can be followed by downstream processing using traditional (alkaline lysis) methods, although NTC has devised an alternative downstream process that uses a temperature inducible autolytic host strain for release of the plasmids from the harvested fermentation cell paste [32,33]. The autolytic process reduces chemical usage in the lysis step by 95%, and costs by 90% – important factors in scaling up, and for reducing the environmental impact of future commercial plasmid manufacturing. It also avoids denaturation-renaturation, which can have unintended consequences on the final product.
Current non-viral vector R&D at NTC involves collaboration with colleagues at institutions that are developing gene therapeutic and immunological products using NTC vectors and technologies. In addition, we are collaborating with the Harbottle Lab at the German Cancer Research Center (Heidelberg, Germany) on the next phase of vector development, combining Nanoplasmids™ and episomally replicating S/MAR vectors [34,35] for improved non-integrative episomal replication vectors.
Acknowledgement
The authors thank Kris Hodgson for artwork.
FINANCIAL & COMPETING INTERESTS DISCLOSURE
The authors are each equity owners of Nature Technology Corporation. No writing assistance was utilized in the production of this manuscript.
References
1. Clinicaltrials.gov; Website
2. Clinical trials worldwide; Website
3. Wahren B, Liu, MA. DNA Vaccines: Recent Developments and the Future. Vaccines 2014; 2(4): 785–96.
CrossRef
4. Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat. Biotechnol. 2000; 18(1): 33–7.
CrossRef
5. Flingai S, Czerwonko M, Goodman J, Kudchodkar SB, Muthumani K, Weiner DB. Synthetic DNA vaccines: improved vaccine potency by electroporation and co-delivered genetic adjuvants. Front. Immunol. 2013; 4: 354.
CrossRef
6. Sullivan SM, Doukas J, Hartikka J, Smith L, Rolland A. Vaxfectin: a versatile adjuvant for plasmid DNA- and protein-based vaccines. Expert Opin. Drug Deliv. 2010; 7(12): 1433–46.
CrossRef
7. Lachish-Zalait A, Lau CK, Fichtman B et al. Transportin mediates nuclear entry of DNA in vertebrate systems. Traffic 2009; 10(10): 1414–28.
CrossRef
8. Dhanoya A, Wang T, Keshavarz-Moore E, Fassati A, Chain BM. Importin-7 mediates nuclear trafficking of DNA in mammalian cells. Traffic 2013; 14(2): 165–75.
CrossRef
9. Lu J, Zhang F, Xu S, Fire AZ, Kay MA. The extragenic spacer length between the 5′ and 3′ ends of the transgene expression cassette affects transgene silencing from plasmid-based vectors. Mol. Ther. 2012; 20(11): 2111–9.
CrossRef
10. Chen ZY, He CY, Kay MA. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum. Gene Ther. 2005; 16(1): 126–31.
CrossRef
11. Williams JA. Improving DNA vaccine performance through vector design. Curr. Gene Ther. 2014; 14(3): 170–89.
CrossRef
12. Williams JA, Luke J, Johnson L, Hodgson C. pDNAVACCultra vector family: high throughput intracellular targeting DNA vaccine plasmids. Vaccine 2006; 24(21): 4671–6.
CrossRef
13. Luke J, Carnes AE, Hodgson CP, Williams JA. Improved antibiotic-free DNA vaccine vectors utilizing a novel RNA based plasmid selection system. Vaccine 2009; 27(46): 6454–9.
CrossRef
14. Luke JM, Vincent JM, Du SX et al. Improved antibiotic-free plasmid vector design by incorporation of transient expression enhancers. Gene Ther. 2011; 18(4): 334–43.
CrossRef
15. Lu J, Zhang F, Kay MA. A mini-intronic plasmid (MIP): a novel robust transgene expression vector in vivo and in vitro. Mol. Ther. 2013; 21(5): 954–63.
CrossRef
16. Williams J. Vectors and methods for genetic immunization. US Patent. 2015; 9109012.
17. Su Y, Connolly M, Marketon A, Heiland T. CryJ-LAMP DNA Vaccines for Japanese Red Cedar Allergy Induce Robust Th1-Type Immune Responses in Murine Model. J. Immunol. Res. 2016; 2016: 4857869.
18. Dutton JL, Woo WP, Chandra J et al. An escalating dose study to assess the safety, tolerability and immunogenicity of a Herpes Simplex Virus DNA vaccine, COR-1. Hum. Vaccin. Immunother. 2016; 12(12): 3079–88.
CrossRef
19. Gerdemann U, Katari UL, Papadopoulou A et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol. Ther. 2013; 21(11): 2113–21.
CrossRef
20. Chen J, Jin M, Qiu ZG et al. A survey of drug resistance bla genes originating from synthetic plasmid vectors in six Chinese rivers. Environ. Sci. Technol. 2012; 46(24): 13448–54.
CrossRef
21. Williams J. Replicative Minicircle Vectors with Improved Expression. US Patent Application. 2015; 2015/0275221
22. Williams J. DNA plasmids with improved expression. US Patent. 2017; 9550998.
23. Monjezi R, Miskey C, Gogishvili T et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2016; 31(1): 186–94.
CrossRef
24. Argyros O, Wong SP, Fedonidis C et al. Development of S/MAR minicircles for enhanced and persistent transgene expression in the mouse liver. J. Mol. Med. 2011; 89(5): 515–29.
CrossRef
25. Broll S, Oumard A, Hahn K, Schambach A, Bode J. Minicircle performance depending on S/MAR-nuclear matrix interactions. J. Mol. Biol. 2010; 395(5): 950–65.
CrossRef
26. Schnodt M, Schmeer, M, Kracher, B et al. DNA minicircle technology improves purity of Adeno-associated viral vector preparations. Mol. Ther. Nucleic Acids 2016; 5: e355.
CrossRef
27. Lu J, Williams JA, Luke J, Zhang F, Chu K, Kay MA. A 5′ Noncoding Exon Containing Engineered Intron Enhances Transgene Expression from Recombinant AAV Vectors in vivo. Hum. Gene Ther. 2017; 28(1): 125–34.
CrossRef
28. Williams JA, Luke J, Langtry S, Anderson S, Hodgson CP, Carnes AE. Generic plasmid DNA production platform incorporating low metabolic burden seed-stock and fed-batch fermentation processes. Biotechnol. Bioeng. 2009; 103(6): 1129–43.
CrossRef
29. Carnes AE, Williams JA. Process for plasmid DNA fermentation. US Patent. 2011; 7943377.
30. Williams J, Carnes, AE. E. coli plasmid DNA production. US Patent. 2016; 9487788.
31. Williams JA, Carnes AE. E. coli plasmid DNA production. US Patent. 2016; 9487789.
32. Carnes AE, Hodgson CP, Luke JM, Vincent JM, Williams JA. Plasmid DNA production combining antibiotic-free selection, inducible high yield fermentation, and novel autolytic purification. Biotechnol. Bioeng. 2009; 104(3): 505–15.
CrossRef
33. Williams J, Hodgson, AP, Carnes, AE. E. coli plasmid DNA production US Patent. 2015; 9017966.
34. Wong SP, Argyros O, Harbottle RP. Sustained expression from DNA vectors. Adv. Genet. 2015; 89: 113–52.
CrossRef
35. Wong SP, Harbottle RP. Genetic modification of dividing cells using episomally maintained S/MAR DNA vectors. Mol. Ther. Nucleic Acids 2013; 2: e115.
CrossRef
AFFILIATIONS
Clague P Hodgson, Aaron E Carnes & James A Williams*
Nature Technology Corporation/Suite
103, 4701 Innovation Drive, Lincoln, NE 68521, USA
*Author for correspondence: jim@natx.com

This work is licensed under a Creative Commons Attribution- NonCommercial – NoDerivatives 4.0 International License.