Bridging preclinical to commercial manufacturing in cell therapy: mind the gap
Cell & Gene Therapy Insights 2025; 11(8), 1037–1044
DOI: 10.18609/cgti.2025.162
Translating innovative cell and gene therapy products from bench to bedside remains hindered by significant translational barriers. Developers face a dilemma: prioritize speed-to-clinic via simple, open-manufacturing systems for a first-in-human trial, or speed-to-market through investments in scalable, compliant manufacturing platforms. Reliance on open systems during early manufacturing, while permissible under Phase 1 cGMP guidelines, poses critical sterility and scalability risks that impede the path to commercialization [1]1.US Food & Drug Administration. Guidance for Industry: cGMP for Phase 1 Investigation Drugs. Jul 2008. .
The strategic question becomes: how can we close the gaps between preclinical, process development (PD), and commercial manufacturing to ensure seamless transition while maintaining phase-appropriate practicality and regulatory compliance?
Understanding the CMC, regulatory, & manufacturing requirements at different stages of product life cycle
Preclinical focus centers on demonstrating product safety, toxicity, pharmacokinetics, efficacy, and mechanism of action (MoA) in GLP-accredited facilities [2]2.Code of Federal Regulations. 21 CFR part 58—good laboratory practice for nonclinical laboratory studies. Dec 22, 1978. . Manufacturing is typically small-scale, using open platforms and research-grade reagents. While cheaper, such reagents (e.g., less purified research-grade viral vectors or FBS) complicate comparability studies and hinder transition to cGMP-compliant processes that require GMP-grade viral vectors or clinical-grade, serum-free, or xeno-free medium. Regulators advise using such defined media early as a key risk mitigation strategy to minimize risk of adventitious agents and batch-to-batch variability.
PD supports CMC documentation in Module 3 of the Common Technical Document for INDs in the USA or the EU Investigational Medicinal Product Dossier (IMPD). While the US FDA guidance allows certain exemptions from 21 CFR Part 211 for Phase 1 clinical trials, fundamental GMP principles from 21 CFR Part 210 still apply: use of GMP-grade reagents, standardized procedures, well-defined processes, calibrated and qualified equipment and facilities, trained personnel, phase-appropriate vendor and material qualification, and demonstrated preliminary data on product safety, identity, purity, and potency [3]3.Code of Federal Regulations. 21 CFR part 210—current good manufacturing practice in manufacturing, processing, packaging, or holding of drugs. Sep 29, 1978. [4]4.Code of Federal Regulations. 21 CFR part 211—current good manufacturing practice for finished pharmaceuticals. . As Phase 1 trials primarily demonstrate safety, with efficacy as a secondary endpoint, having flexibility with non-commercial ready, yet robust manufacturing workflows would be most appropriate for the majority of early-stage biotechs. Progression to later trials, however, requires a transition towards closed, scalable workflows and deeper process knowledge to support commercial manufacturing.
Analytics also evolve across stages: early-stage potency assays may qualitatively demonstrate biological activity, but often lack the robustness, specificity, and reproducibility required for lot release, mandated by specifications in ICHQ6B [5]5.European Medicines Agency. ICH Q6B specifications: test procedures and acceptance criteria for biotechnological/ biological products. Sep 1, 1999. . Bridging studies are often needed to compare legacy and improved analytical assays as part of continuous improvement. It is inevitable that early decisions made in the PD stage may have implications for the clinical pipeline development, underscoring the need for a clear roadmap for process upgrade before pivotal trials and commercial manufacturing.
Full cGMP compliance is mandatory by commercial stages. Adopting validated, closed-system workflows with defined proven acceptance range (PAR) and normal operating range (NOR) is a key strategy to mitigate risk during the manufacturing process and enhancing commercial viability. Processes, equipment, and facilities must be validated for scale-up and scale-out to accommodate increasing manufacturing demands. Analytical methods must be developed in line with ICH Q14 guidelines and validated as per ICHQ2 guidance [6]6.European Medicines Agency. ICH Q2(R2) validation of analytical procedures—scientific guideline. Jun 14, 2024. [7]7.European Medicines Agency. ICH Q14 analytical procedure development—scientific guideline. . Materials and vendors must be qualified according to 21 CFR Parts 210 and 211 [3]3.Code of Federal Regulations. 21 CFR part 210—current good manufacturing practice in manufacturing, processing, packaging, or holding of drugs. Sep 29, 1978. [4]4.Code of Federal Regulations. 21 CFR part 211—current good manufacturing practice for finished pharmaceuticals. , US and EU Pharmacopeia ancillary materials standards [8]8.United States Pharmacopeia. General Chapter, <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products. [9]9.European Pharmacopoeia. General Chapter 5.2.12. Raw Materials of Biological Origin for the Production of Cell-Based and Gene Therapy Medicinal Products., and relevant biologics-specific requirements outlined in 21 CFR Parts 600-680 [10]10.Code of Federal Regulations. 21CFR Parts 600–680—biological product manufacturing, licensing, and standards. . Robust procurement, supply chain and cold-chain logistics, chain-of-custody, and data management systems are essential for regulatory approval and inspections (Table 1).
Table 1. Regulatory and CMC requirements by development phase | ||
|---|---|---|
Preclinical phase | Process development/IND | Commercial phase |
Research grade reagents | GMP principles (21CFR210) | Full cGMP (21CFR210-211) |
Open systems | Phase-appropriate controls | Manufacturing process validation |
GLP requirements (21CFR58) | Closed workflows | PAR/NOR set |
Small-scale manufacturing | Valid equipment | ICH Q2/Q14 qualified analytical methods |
Safety/efficacy | Trained staff | Qualified suppliers |
Manual operation | Identity/purity/potency | Supply chain validated |
Streamlining the path from academia to industry
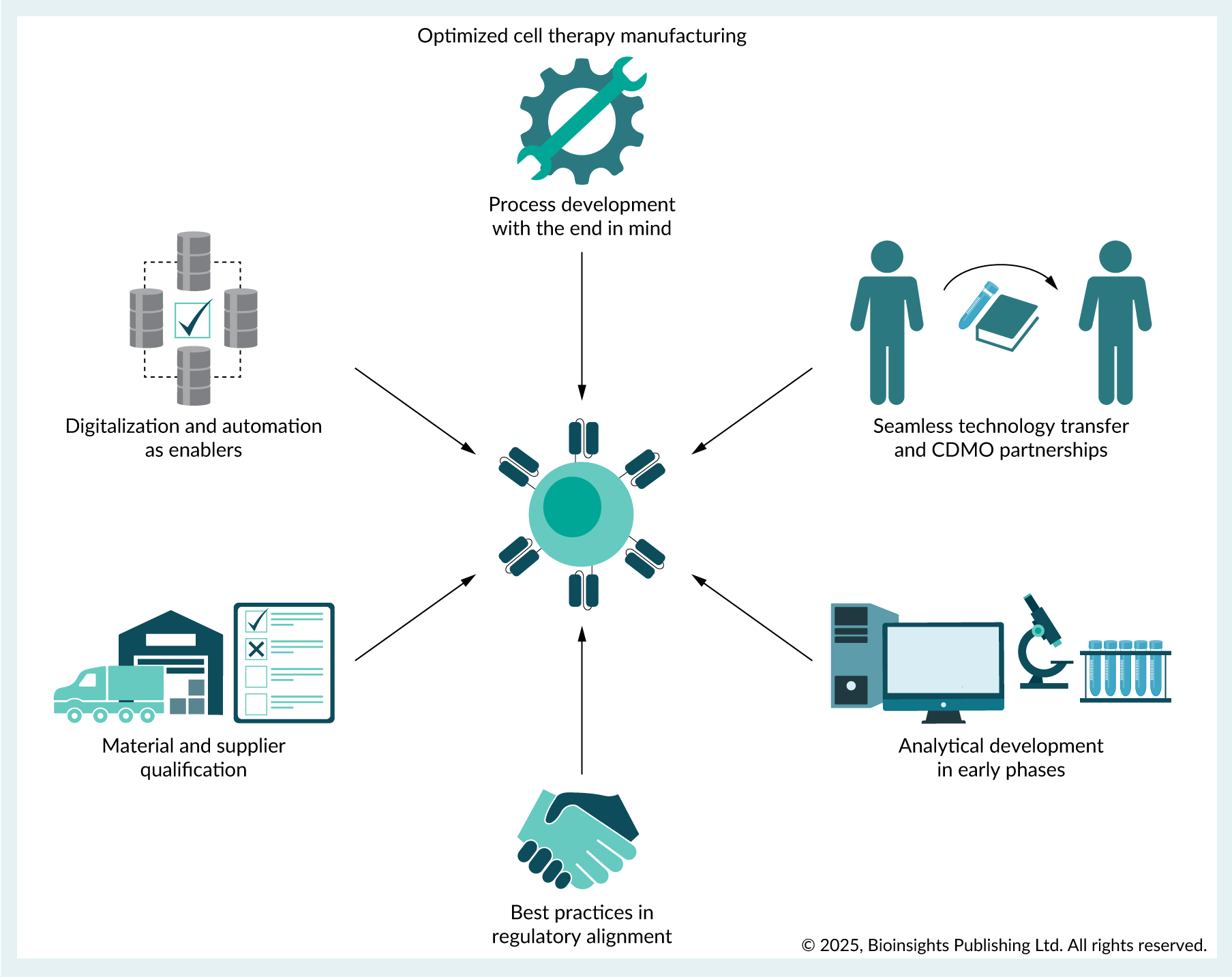
Embedding commercial translation requirements into early PD minimizes costly delays, increasing commercial viability. This ‘begin with the end in mind’ approach requires early alignment on commercial-scale manufacturing strategies such as adoption of automated scale-up or scale-out manufacturing platforms early in development. Integration of digitalization during development eases future manufacturing and streamline data collection. Future-proofing analytical capabilities by incorporating Process Analytical Technologies (PAT) for real-time monitoring during manufacturing enhances product quality control (Figure 1).
| Figure 1. Early adoption of best practices for commercially aligned cell therapy manufacturing. |
|---|
 |
| © 2025, BioInsights Publishing Ltd. All rights reserved. |
Process development with end in mind: QbD
Implementing QbD principles early in PD using multivariate and DoE methodology allows to define the process design spaces and link critical process parameters (CPPs) with critical quality attributes (CQA). Early mapping and control of CPPs via close collaboration between preclinical and PD teams streamlines the transition towards process validation and Process Performance Qualification (PPQ) in later manufacturing phases.
Analytical development in early phases
Analytical methods must co-evolve with process understanding. Early adoption of PAT enables real-time monitoring and adaptive control, while establishing a matrix approach to assay development comprehensively captures product potency and MoA for regulatory submissions [11]11.US Food & Drug Administration. GuidanceDocument. Potency Assurance for Cellular and Gene Therapy Products. Dec 2023. . These approaches facilitate the integration of machine learning and AI tools aligned with Manufacturing 4.0 principles to enhance product quality assessment and release.
Closed system manufacturing, scalability, & automation
Early integration of phase-appropriate processes minimizes costly downstream comparability and bridging studies. Harmonizing early-stage development with commercialization demands adopting processes with the end in mind. Utilization of closed-system automated manufacturing platforms that support autologous and allogeneic workflows, offer integrated cell processing and expansion, and can be validated for compliance with 21 CFR Part 11 requirements, facilitating a smoother transition towards commercial scale-out manufacturing. PD work on these platforms establishes a clearer path towards commercial manufacturing without necessitating late-stage comparability studies that regulatory authorities may require if platform changes occur later. Interconnectivity across different manufacturing equipment and digital control systems is equally important for modular manufacturing workflows and can be achieved with software solutions that provide end-to-end data integration and orchestration.
Adoption of FDA advanced manufacturing technology
Early adoption of automated manufacturing platforms with FDA Advanced Manufacturing Technology (AMT) designation [12]12.US Food & Drug Administration. Guidance Document. Advanced Manufacturing Technologies Designation Program. Dec 2024. can accelerate the transition towards regulatory approvals. These can include AI-driven platforms for automated, scalable manufacturing of iPSCs, and large-scale, self-contained ‘GMP-in-a-box’ systems designed for industrial production of immune cell therapies. Innovative, compact benchtop solutions utilizing microfluidics or other modular approaches provide cGMP-compliant, end-to-end solutions that enable a seamless transition of products from pre-clinical research through to commercial-scale manufacturing.
Leveraging external expertise for manufacturing
Early partnerships with CDMOs leverage their PD and commercial manufacturing expertise to accelerate commercialization. Many CDMOs now offer access to established, therapy-class-specific manufacturing platforms that leverage expertise developed across multiple client pipelines, significantly reducing development timelines. This advantage stems from pre-existing master batch records, validated standard operating procedures (SOPs), qualified analytical methods, completed aseptic validation, experienced personnel, and reliable supply chains. Partnering with such CDMOs offering pre-optimized platforms minimizes upfront PD investments, expedites commercial development, reduces manufacturing failure risk, and lowers costs for IND-enabling studies and clinical production.
Supply chain management
Validated supply chains are needed to ensure that high-quality raw materials and starting materials are available for manufacturing. Having established and reliable supplier relationships ensures stable and continuous supply of ancillary raw materials for disruption free manufacturing. Identification of alternate materials and their impact on safety, quality, purity, identity, potency and stability of the manufactured product mitigates supply chain risks, ensuring continuity of production and avoiding requirements for comparability studies that may arise due to inadvertent late stage change in raw material utilization. Procedures to identify, establish, and periodically review primary and alternate suppliers should be implemented during early-stage clinical development and are useful for transition towards commercial manufacturing. Validated collection and shipping of healthy or patient starting source materials for manufacturing, chain-of-custody management, and subsequent shipping of manufactured product for clinical administration would be critical for clinical operations.
Digitalization
Another important area for early adoption is the shift from laborious and tedious paper-based records towards digital solutions for establishing quality management systems (QMS), manufacturing batch records, eSOP, change and deviation management, inventory, workflow scheduling management, and supply-chain tracking, in a manner that is compliant with 21 CFR Part 11 [13]13.US Food & Drug Administration. Guidance Document. Part 11, Electronic Records, Electronic Signatures. Sep 2003. . This modernization can be supported by a variety of specialized digital tools, including dedicated software for production planning and resource scheduling; comprehensive cell therapy orchestration platforms providing end-to-end workflow management with chain of identity and custody tracking; and enterprise-level eQMS for overseeing documentation, deviations, and change control. When combined with digital interconnectivity of manufacturing equipment, these digital workflow management solutions enable facilities to improve productivity, while optimizing facility and resource utilization.
Best practices in regulatory alignment
Fostering a collaborative and transparent relationship with regulatory agencies is another key de-risking strategy to streamline progress towards commercialization: leveraging early engagement opportunities such as the FDA’s INTERACT meetings (USA), the EMA’s Innovation Task Force briefings and Scientific Advice procedures (EU), MHRA’s ILAP (UK) and PMDA Sakigake (Japan). Such discussions allow for early feedback on CMC strategy, proposed analytical panel, or overall development plan, helping to align expectations and build regulatory confidence in the program.
Conclusion
A phase-appropriate approach for cell therapy manufacturing is essential, but commercial foresight from day one greatly increases the odds of success. Early adoption of enabling technologies, QbD methodology, automation, digitalization, and CDMO partnerships can ensure that a cell therapy program moves efficiently from preclinical, early-stage trials through to commercial manufacturing, minimizing the risk of costly bottlenecks or late-stage redesigns. Embedding these essential best practices strategically into phase appropriate development will improve the likelihood that cell therapy pipelines achieve both timely patient access and sustainable scalability for commercial success.
References
1. US Food & Drug Administration. Guidance for Industry: cGMP for Phase 1 Investigation Drugs. Jul 2008. Link
2. Code of Federal Regulations. 21 CFR part 58—good laboratory practice for nonclinical laboratory studies. Dec 22, 1978. Link
3. Code of Federal Regulations. 21 CFR part 210—current good manufacturing practice in manufacturing, processing, packaging, or holding of drugs. Sep 29, 1978. Link
4. Code of Federal Regulations. 21 CFR part 211—current good manufacturing practice for finished pharmaceuticals. Link
5. European Medicines Agency. ICH Q6B specifications: test procedures and acceptance criteria for biotechnological/ biological products. Sep 1, 1999. Link
6. European Medicines Agency. ICH Q2(R2) validation of analytical procedures—scientific guideline. Jun 14, 2024. Link
7. European Medicines Agency. ICH Q14 analytical procedure development—scientific guideline. Link
8. United States Pharmacopeia. General Chapter, <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products. Link
9. European Pharmacopoeia. General Chapter 5.2.12. Raw Materials of Biological Origin for the Production of Cell-Based and Gene Therapy Medicinal Products. Crossref
10. Code of Federal Regulations. 21CFR Parts 600–680—biological product manufacturing, licensing, and standards. Link
11. US Food & Drug Administration. GuidanceDocument. Potency Assurance for Cellular and Gene Therapy Products. Dec 2023. Link
12. US Food & Drug Administration. Guidance Document. Advanced Manufacturing Technologies Designation Program. Dec 2024. Link
13. US Food & Drug Administration. Guidance Document. Part 11, Electronic Records, Electronic Signatures. Sep 2003. Link
Biographies
Ashwin Srinivasan Kumar is a translational scientist with 8 years of experience focused on advancing cell therapies from the lab to the clinic. In his current role at A*STAR’s Process Accelerator for Cell Therapy Manufacturing, he is responsible for developing robust manufacturing processes for novel immunotherapies. Dr Kumar’s research leverages state of the art technologies to dissect the complex tumor immune microenvironment. His technical expertise includes high-throughput screening, single-cell genomics, advanced bio-imaging, computational modeling, and finite element analyses to identify novel therapeutic targets and biomarkers. He has a proven track record of translating complex biological questions into robust, data-driven experimental designs, aiming to bridge the gap between discovery and clinical application. His current work builds on his postdoctoral research at Harvard Medical School and the Broad Institute, which focused on translating novel immunotherapies for pediatric medulloblastoma. Dr Kumar earned his PhD in Medical Engineering and Medical Physics from MIT, Cambridge, MA, USA and a BEng from Imperial College London, UK. He has authored multiple peer-reviewed publications in leading scientific journals.
Jaichandran Sivalingam is a Principal Scientist and Group Leader for the Process Accelerator for Cell Therapy Manufacturing (PACTMAN) group at Bioprocessing Technology Institute, A*STAR. A molecular biologist by training with over 20 years of research and industry experience in cell and gene therapy development. He was previously involved in pre-clinical cell therapy pipeline development for diabetes and hemophilia A, at the National Cancer Centre, Singapore. His expertise in cell therapy development includes gene engineering and establishing scalable manufacturing of induced pluripotent stem cells derived hematopoietic stem cells and universal red blood cells for transfusion therapy at BTI (A*STAR). He was part of the Process, Science and Technology (PSAT) team involved in CAR-T pipeline development at Tessa Therapeutics, formerly a late-stage clinical company involved in immune-oncology pipeline development. He currently leads the PACTMAN group, A*STAR’s cell and gene therapy initiative, to provide pre-GMP Process and Analytical Development support for cell therapy asset developers from academia, national healthcare groups and local biotech companies and serves as a review editor on the editorial board of Cancer Immunity and Immunotherapy.
Ashwin Srinivasan Kumar and Jaichandran Sivalingam, Process Accelerator for Cell Therapy Manufacturing, Bioprocessing Technology Institute, Agency for Science, Technology and Research (A*STAR), Singapore
Sabry Hamza is a seasoned biotechnology leader with over 20 years of experience spanning academia and industry, specializing in virology, oncology, immunology, neuroscience, and cell biology. He currently serves as Head of Process Development and Analytical Sciences at Tikva Allocell, where he leads the strategic design, development, and optimization of CAR-T cell manufacturing processes and analytical methods for cell and gene therapies. Dr Hamza has a proven track record of guiding programs from early discovery through clinical development, including drafting Chemistry, Manufacturing, and Controls (CMC) sections for IND and global regulatory submissions. His expertise includes establishing scalable manufacturing workflows, developing lot release assays, and ensuring regulatory compliance with FDA and EMA guidelines. Prior to his current role, he held senior positions at Tessa Therapeutics and Merck Sharp & Dohme, where he contributed to innovative platforms in cell therapy and translational medicine research. Dr Hamza earned his PhD in Comparative Pathology from the University of California, Davis, CA, USA and his BS in Biological Sciences from California State University, Chico, CA, USA. He has authored multiple peer-reviewed publications and serves as an editorial board member and scientific reviewer in oncology and integrative cancer research.
Sabry Hamza, TIKVA Allocell, Singapore
Authorship & Conflict of Interest
Contributions: The named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors have no conflicts of interest.
Funding declaration: The authors received no financial support for the research, authorship and/or publication of this article.
Article & Copyright Information
Copyright: Published by Cell & Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2025 Ashwin Srinivasan Kumar, Sabry Hamza, Jaichandran Sivalingam. Published by Cell & Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited.
Revised manuscript received: Aug 29, 2025.
Publication date: Sep 18, 2025.